Projetos em andamento

|
Abordagens mult-ômicas para estudo de comunidades microbianas para descoberta de novas estratégias de desconstrução de biomassa lignocelulósica (lnbr_bag19) Centro Nacional de Pesquisa em Energia e Materiais Laboratório Nacional de Biorrenováveis Coordenador: Gabriela Felix Persinot Áreas do conhecimento: Biodiversidade; Ciências biológicas Início da vigência: 15-10-2019 A biomassa lignocelulósica é a principal fonte de carboidratos renováveis do mundo e é considerada uma plataforma sustentável para a produção de bioprodutos como biocombustíveis, blocos químicos de base biológica e biopolímeros. Um dos principais desafios da bioconversão de biomassa lignocelulósica em bioprodutos é a transformação dos polissacarídeos presentes na biomassa em açúcares fermentescíveis. Comunidades microbianas presente em ambientes naturais como solos recoberto por biomassa e intestino de aninais herbívoros como ruminantes, possuem naturalmente grande capacidade de obter energia a partir de biomassa lignocelulósica. Essa capacidade encontra-se codificada no potencial genético dos micro-organismos presentes nestes ambientes, porém, muitas vezes esse potencial é pouco ou apenas superficialmente explorado. O recente desenvolvimento das tecnologias de sequenciamento de ácidos nucleicos, impulsionaram o uso de abordagens em larga escala independentes de cultivo, como a metagenômica, a qual se tornou uma poderosa ferramenta que permite acessar o potencial genético de comunidade microbianas por meio do sequenciamento direto do DNA ambiental. Neste sentido, a metagenômica é uma técnica promissora para prospecção de enzimas degradadoras de biomassa vegetal, pois além de permitir estudos de diversidade microbiana, permite acessar o genoma desses organismos incultiváveis possibilitando a descoberta de genes envolvidos em variadas atividades enzimáticas de sistemas biológicos até então desconhecidos. Estas técnicas apenas fazem sentido se suportados pela área de pesquisa de Bioinformática e computação de alto desempenho, as quais visam atribuir significado biológico ao enorme volume de dados gerado. Nesse sentido, este projeto visa o desenvolvimento e aplicação de abordagens in silico de Bioinformática para explorar o potencial genético de comunidades microbianas de forma a aprofundar o conhecimento acerca da diversidade de micro-organismos e enzimas presentes em ambientes naturais, visando compreender as estratégias naturalmente empregadas pelas comunidades microbianas para desconstrução de materiais lignocelulósicos e explorar de forma racional o potencial existente para a prospecção de novas enzimas ou microrganismos. Por meio de análises integradas multi-ômicas de comunidades microbianas recuperada de solo recoberto com bagaço de cana-de-açúcar e de intestino de herbívoros, espera-se com esse projeto poder contribuir com a elucidação da diversidade filogenética dessas comunidades e com isso possibilitar a descoberta de genes, enzimas, genomas e microrganismos envolvidos na degradação de materiais lignocelulósicos, que podem ser de grande interesse para aplicações biotecnológicas. |
|
|
Análise Computacional de proteínas consideradas alvos moleculares para o tratamento da esquistossomíase e leishmanioses (nddrug) Universidade Federal de Juiz de Fora Departamento de Ciência da Computação Coordenador: Priscila Vanessa Zabala Capriles Goliatt Áreas do conhecimento: Ciência da computação; Ciências biológicas; Ciências da saúde; Farmácia; Química Início da vigência: 10-07-2017 Doenças parasitárias são um dos principais problemas de Saúde Pública, porém são consideradas como doenças negligenciadas por serem endêmicas em populações de baixa renda e por apresentarem investimentos reduzidos em produção de medicamentos. Como exemplo, pode-se citar a esquistossomíase (ou esquistossomose) e as leishmanioses. A esquistossomíase é uma doença aguda ou crônica que acomete principalmente o intestino. É causada por vermes do gênero Schistosoma, destacando-se o S. mansoni, e afeta cerca de 239 milhões de pessoas no mundo, com cerca de 6 milhões de pessoas infectadas somente no Brasil. As leishmanioses, causadas por protozoários do gênero Leishmania, possuem manifestações desde lesões cutâneas até injúrias severas das vísceras, podendo levar a óbito. O CDC (do inglês, Centers for Disease Control and Prevention) considera as leishmanioses como a segunda maior causa de morte no mundo por doenças parasitárias, sendo reportados cerca de 2 milhões de novos casos/ano. As drogas usuais possuem baixa eficácia e diversos efeitos colaterais devido esses parasitas apresentarem enzimas com funções semelhantes às encontradas em seres humanos. Adicionalmente, é crescente o número de eventos de resistência a essas drogas. Portanto, a busca por enzimas ditas potencias alvos moleculares é fundamental para o desenvolvimento de drogas mais eficazes e com menos efeitos colaterais, promovendo maior adesão dos pacientes ao tratamento completo dessas doenças. Os altos custos dos estudos experimentais têm elevado o uso de ferramentas de Biologia Computacional para entender a estrutura de alvos moleculares e os mecanismos de interação entre essas proteínas e seus possíveis inibidores. O modo de interação entre essas proteínas em S. mansoni e Leishmania sp. e compostos com potencial inibitório ainda é desconhecido. Em estudos anteriores, realizamos a modelagem tridimensional da NTPDase1 de S. mansoni (SmNTPDase1), enzima considerada alvo molecular para o tratamento da esquistossomíase, e seu homólogo em humanos (HsNTPDase1 ou CD39). Através da análise comparativa entre a SmNTPDase1 e a NTPDase1, por dinâmica molecular (DM) e docking com compostos candidatos a inibidores da SmNTPDase1, verificamos que mudanças conformacionais importantes nessas proteínas levam a diferentes afinidades de interação com os inibidores. Através deste projeto, poderemos elucidar esses modos de interação, de modo a auxiliar na remodelagem desses compostos ou no desenho racional de novos fármacos mais eficazes e com menos efeitos colaterais. Atualmente, alvos em L. braziliensis e L. amazonensis estão sendo mapeados. Alvos como a tripanotiona reductase já foram modelados, estão sendo testados contra compostos candidatos à leishmanicida e deverão seguir para análises por DM. Em computadores com arquitetura multicore, as análises por DM de cada alvo podem custar até um ano de simulação. Para a análise comparativa dessas enzimas e seus homólogos humanos esse custo pode inviabilizar um projeto com financiamento de até 24 meses. Portanto, é necessário o uso de supercomputadores com GPU, sendo o SDumont um componente essencial neste projeto. Acreditamos que este projeto possa impactar na validação de compostos em estudo como antiparasitários, culminando na elaboração de patentes. Acreditamos também, que nossa pesquisa tem impacto científico relevante, cujos resultados podem representar contribuições biotecnológicas diretas em setores produtivos da sociedade. |
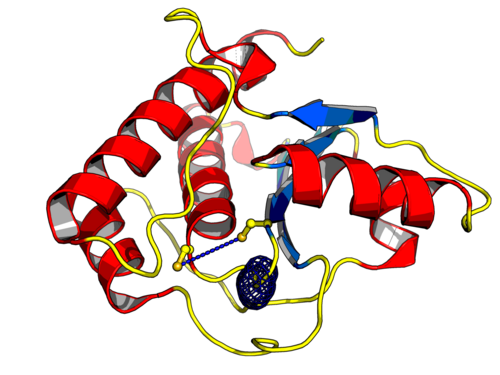
|
Análise de mutações correlacionadas por Dinâmica Molecular em fosfatases de baixo peso Molecular (lmwptpdm) Universidade Federal de Minas Gerais Instituto de Ciências Biológicas Coordenador: Lucas Bleicher Áreas do conhecimento: Ciências biológicas Início da vigência: 27-07-2016 Um dos eventos celulares de mediação química mais importantes e úbiquos nos organismos vivos é a fosforilação, cuja dinâmica ocorre devido a existência de múltiplas enzimas responsáveis por catalisar a adição (quinases) e remoção (fosfatases) de grupos fosfato em outras moléculas. Em organismos multicelulares, a fosforilação/desfosforilação de Tirosina em proteínas é especialmente importante para diversas vias de sinalização celular. Dentre os diversos ramos da ciência mundial, a Bioinformática é tida como uma área de desenvolvimento recente e de grande interdisciplinariedade, sendo imprecindível ao tratamento adequado das grandes quantias de dados biológicos obtidas pelas técnicas de Biologia Molecular atuais, assim sendo capaz de resolver inúmeros problemas de natureza biológica. Nesse estudo, utilizamos inicialmente técnicas estatísticas para a extração de possíveis correlações e anticorrelações entre amino ácidos de um alinhamento correspondente a uma família ubíqua e ancestral de tirosino-fosfatases: As Fosfatases de Baixo Peso Molecular (do inglês LMW-PTPs). Esta família de proteínas, agrupa enzimas de um padrão único de enovelamento, mas que diferem em um um grupo capaz de desfosforilar tirosina e outro cujas enzimas são responsáveis pela redução de Arsenato em Arsenito. As LMW-PTPs humanas são relacionadas a múltiplas doenças, das quais destacam-se inclusive alguns tipos de câncer. Ainda não se entende completamente quais os mecanismos moleculares que implicam em diferenças na especificidade e na atividade das proteínas desta família. Inicialmente buscamos relacionar funções biológicas a grupos de resíduos correlacionadas através de uma revisão bibliográfica e de análises em estruturas cristalográficas desta família. Estas evidenciaram o papel de vários resíduos de nossa análise, embora as funções biológicas relacionadas a algumas de nossas correlações sejam bastante elusivas. Existe portanto interesse científico na descrição dos mecanismos moleculares de especificidade, nos processos alostéricos dessas enzimas, e no desenvolvimento de possíveis inibidores as mesmas. O estudo aqui proposto destas enzimas, e do papel dos resíduos que encontramos neste trabalho por técnicas de Dinâmica Molecular será então capaz de promover um avanço no entendimento dessa família de proteínas, podendo servir como base a futuros ensaios experimentais, além de ser capaz de divulgar e gerar conhecimento científico em uma área de ponta. |
|
|
Atomistic Simulations of Material’s interfaces for solar energy devices (asmsol) Universidade Estadual Paulista Júlio de Mesquita Filho Instituto de Física Teórica Coordenador: Alexandre Reilly Rocha Áreas do conhecimento: Química Início da vigência: 28-09-2016 This project aims to provide accurate predictions at atomic and molecular level of the interfaces of solar energy devices, which would be used for experimentalist in the seeking of new materials for designing novel solar energy devices. Our research proposal involves state of art simulations, which play a key role in predicting physical and chemical properties at atomistic scale, for studying the charge-transfer (CT) processes occurring at the interfaces of OPCs. Special attention will be given in seeking different blend of donor/acceptor materials based: on graphene nanoribbons and organic semiconductors. In particular, we will tailor the energy levels of GNRs by functionalization and doping, in order to explore its role over the CT states to obtain heterojunctions with improved properties. To this aim, the methodology will be based on the many-body perturbation theory, which have been successfully used in describing excited states of several molecular systems. Thus, within the so-called GW approximation, we will be able to correct the Kohn-Sham (DFT) energy levels at the interfaces of the blend of donor/acceptor. Furthermore, by using these corrected energies and by solving the Bethe-Salpeter equation (BSE), we will be able to simulate accurately the optical properties, energy-loss spectra and the charge-transfer process at the interfaces of OPCs as well as their implications for designing more efficient devices. We also want to understand the electrodes, which are used to harvest the electrons and holes generated by light adsorption. In this case we need to understand the transport properties of this materials, which typically contain impurities. Understanding the effects of these impurities is tantamount for the design of good electrodes. |
|
|
Balanceador de carga distribuído MPI para métodos geofísicos com grandes instâncias de dados (ctws-fwi) Universidade Federal do Rio Grande do Norte Departamento de Engenharia de Computação e Automação Coordenador: Samuel Xavier de Souza Áreas do conhecimento: Ciência da computação; Engenharias; Geociências Início da vigência: 07-06-2019 O balanceamento centralizado de grandes cargas de trabalho pode fazer com que o escalonamento se torne um gargalo de desempenho e eficiência. Cada vez maiores em tamanho, levantamentos sísmicos 3D requerem uma logística de dados cada vez mais complexa para distribuir dados de tiros para os nós de computação em ambientes HPC. A fim de manter o tempo dos resultados dentro dos limites desejáveis, muitas vezes a decomposição do domínio dentro de um único tiro também é aplicada, tornando a pressão sobre um escalonador centralizado ainda maior. Este projeto tem como objetivo desenvolver um escalonador descentralizado para balancear grandes cargas de processamento em grandes infraestruturas de HPC. Serão empregadas técnicas como o "work stealing" que já foram aplicadas com sucesso para equilibrar a carga de trabalho do Cilk Plus, um modelo de programação de memória compartilhada desenvolvido no MIT e que agora pertence à Intel Software. A responsabilidade do balanceamento de carga deve ser distribuída entre os nós computacionais sem uma autoridade de escalonamento centralizada para evitar agravar gargalos computacionais. Ao longo da evolução da computação, tarefas podem ser trocadas entre nós para evitar que processadores fiquem ociosos. Embora a técnica proposta possa ser aplicada a diversos métodos computacionais, nossas aplicações iniciais serão a Migração Reversa no Tempo (RTM, do inglês Reverse Time Migration) e a Inversão Completa da Forma de Onda (FWI, do inglês Full Waveform Inversion). |
|
|
Brazilian Earth System Model: seasonal to centenial climate prediction (BESM-S2C) Instituto Nacional de Pesquisas Espaciais Centro de Previsão do Tempo e Estudos Climáticos Coordenador: Paulo Nobre Áreas do conhecimento: Ciência da computação; Ciências biológicas; Matemática; Meteorologia; Oceanografia; Probabilidade e estatística; Química Início da vigência: 07-03-2017
|
|
|
Buscando o mínimo global dos métodos quânticos semiempíricos NDDO (rm2) Universidade Federal de Pernambuco Departamento de Química Fundamental Coordenador: Alfredo Mayall Simas Áreas do conhecimento: Química Início da vigência: 15-07-2016
|
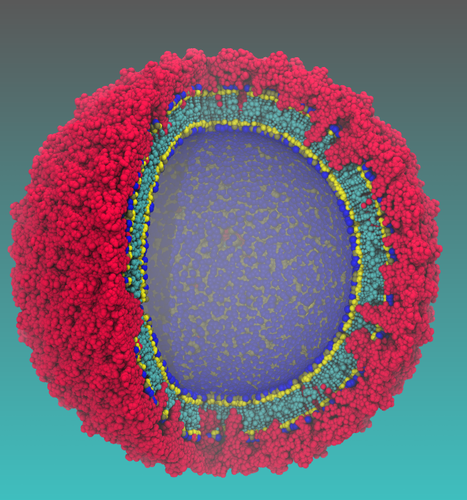
|
Caracterização in silico de alvos de medicamentos para Zika e Dengue (VICBF01) Universidade Federal de Pernambuco Departamento de Química Fundamental Coordenador: Gustavo de Miranda Seabra Áreas do conhecimento: Ciências biológicas; Química Início da vigência: 08-11-2016 Este é um trabalho sendo desenvolvido em colaboração com o prof. Sergio Pantano, do Group of Biomolecular Simulations, Institut Pasteur de Montevideo (Uruguay). A atual epidemia de Zika e outros flavivírus no Brasil e na América Latina aumentou em todo mundo a preocupação da saúde publica por causa das implicações médicas e sócio econômicas. Consequentemente, um grande esforço começou a ser dedicado para enfrentar esse problema. Nesses contexto, simulações de dinâmica molecular compõem uma importante alternativa para prover informações confiáveis para o Design racional de ferramentas terapêuticas. Além disso, instalações de supercomputadores permitem a triagem de um grande número de condições (i.e., mutações, concentrações, temperatura, pH, etc.) ajudando na aceleração, racionalização ou até mesmo evitando o consumo excessivo de tempo de trabalho experimental. Neste projeto nos propomos um colaboração internacional com foco em dois alvos de design de drogas: i) A proteína NS3 protease/helicase; e ii) a proteína de envelope. A proteína NS3 do vírus Zika possui grande identidade de sequencia com a mesma proteína em outros flavivirus, como o Dengue. Essa proteína possui dois domínios, protease e helicase, e é essencial no processo de replicação viral. Na realidade, alguns medicamentos antivirais já existentes no mercado, para HIV e Hepatite C, se baseiam na inibição da atividade desta proteína. Pretendemos utilizar o programa PMEMD, do pacote Amber16, para simular as etapas envolvidas na função da NS3 helicase, responsável pelo desenovelamento do RNA viral, para identificar sítios passíveis de ataque. Quanto ao domínio protease, pretendemos estudar a reação catalisada por esse domínio através de cálculos QM/MM, para identificar o mecanismo e as estruturas importantes, que podem também oferecer alvos para novos medicamentos. A segunda proteína é também um dos principais alvos, assim como a primeira no reconhecimento pela célula hospedeira. Recentemente, nós desenvolvemos e aplicamos um método de simulação Coarse Grained (Darré L, Machado MR, Brandner AF, Ferreira S, Gonzalez HC, Pantano S. SIRAH: a structurally unbiased coarse-grained force field for proteins with aqueous solvation and long-range electrostatics. JCTC, 2015, 11:723.) proposto de multi-escala para estudar a flexibilidade de estruturas recém-publicadas do envelope do Zika (Sirohi, D.; Chen, Z.; Sun, L.; Klose, T.; Pierson, T. C.; Rossmann, M. G.; Kuhn, R. J. Science The 3.8 A resolution cryo-EM structure of Zika virus: 2016, 352, 467-470). A introdução de mutações pontuais na estirpe Africana revela a estabilização estrutural do envelope do vírus (Machado et al, submetido ao JCTC), que acontece apenas a nível da estrutura quaternária. Nós usaremos a estrutura do vírus do Zika junto com a modelagem molecular e simulações multi-escala para gerar um conjunto completo de estruturas (aproximadamente 50 diferentes variantes com identidade ~90%, http://www.ianlogan.co.uk/zikapages/zika-current.htm). Isto proverá informações preciosas sobre a acessibilidade dos epítopes e sua estrutura, bem como a variabilidade dinâmica de diferentes variantes que circulam no Brasil. Nós planejamos gerar 1 microssegundo de simulações de dinâmica molecular de cada variante para gerar um imprescindível banco de dados que será disponibilizado para a comunidade cientifica por meio de websites com o objetivo de ajudar os biólogos computacionais que trabalham ao lado dos experimentalistas para antecipar o impacto de novas mutações. |
|
|
Derivação, otimização e implementação de parâmetros para boro, estanho e platina nos campos de força MMFF94S e AMBER, com potencial para o desenho de novas drogas (mmffdbsnpt) Universidade Federal Fluminense Instituto de Química Coordenador: Luciano Tavares da Costa Áreas do conhecimento: Ciências biológicas; Química Início da vigência: 12-07-2018 Compostos químicos contendo Pt , B ou Sn exibem potencial para fármacos antineoplásicos. Diversos complexos de Pt têm sido estudados como agentes antiproliferativos desde a aprovação do uso clínico da cisplatina, cis-[PtCl2(NH3)2], pelo FDA (Food and Drug Administration), no final da década de 1970. Cisplatina ainda é, hoje em dia, o principal anticancerígeno com Pt. Também é crescente o interesse na atividade biológica de moléculas com boro. Elas têm sido investigadas com o agentes anticancerígenos e antimicrobianos. Bortezomib é um medicamento antineoplásico contendo um ácido borônico , [R-B(OH)2], como princípio ativo. Alguns complexos de Sn exibem atividade antiproliferativa maior que aquela observada para cisplatina ou outras drogas inorgânicas. Por isso, a procura por a gentes anticancerígenos com Sn, sobre tudo organoestânicos(IV), tem crescido nas últimas décadas. Fármacos com Pt, B e Sn exibem, geralmente, vários efeitos colaterais. Portanto, ainda existe a necessidade do desenvolvimento de fármacos que causem menos efeitos secundários e consigam contornar mecanismos de resistência tumoral. O desenho racional e o desenvolvimento de novas drogas utilizam-se do atracamento molecular (docking), que busca avaliar a afinidade de um ligante pelo sítio de ligação da proteína-alvo, seja ele ortostérico ou alostérico. DockThor é um programa de atracamento molecular com portal web acoplado ao uso do supercomputador SDumont para triagem virtual em larga escala (http://www.dockthor.lncc.br). Ele adota uma função de avaliação baseada no campo de força MMFF9 4 S. Nele, não há parâmetros para moléculas com Pt, B ou Sn. Este projeto, portanto, visa à obtenção de parâmetros intra e intermoleculares, incluindo constantes de força e cargas atômicas, baseados nesses elementos em compostos biologicamente ativos, empregando cálculos HF, MP2 e DFT, e à implementação desses parâmetros nos campos de força MMFF9 4 S e AMBER. Esta proposta objetiva o desenvolvimento de uma versão do DockThor capaz de avaliar poses para compostos de Pt, B ou Sn, ampliando a sua capacidade preditiva, e aprovisão de parâmetros que permitam simulações clássicas usando AMBER. Nesta proposta, visando à derivação de novos parâmetros para ambos campos de força, está prevista a análise de cerca de 2700 moléculas. Isso viabilizaria o uso conjunto de docking e DM para o desenho de novas drogas com Pt, B e Sn. |
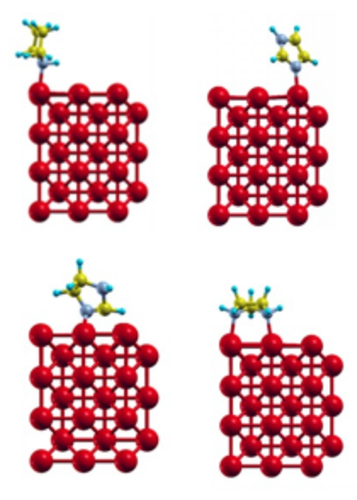
|
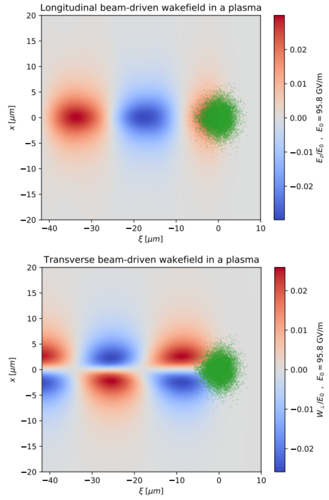
Desaceleração de feixes de partículas carregadas por campo elétrico de rastro (bdwf) Universidade Federal do Rio Grande do Sul Departamento de Engenharia Elétrica Coordenador: Roger Pizzato Nunes Áreas do conhecimento: Física Início da vigência: 19-07-2019 A técnica de aceleração de partículas carregadas por indução de um campo de rastro em um plasma representou um dos maiores avanços na área de feixes de partículas carregadas. A possibilidade de acelerar feixes a energias da ordem dos GeV em apenas alguns centímetros no canal de aceleração permitiu o projeto e o desenvolvimento de máquinas mais potentes e compactas. A potência é um fator importante em aplicações científicas dos feixes de partículas carregadas, tais como investigação da estrutura da matéria, enquanto a compacticidade é relevante nas aplicações na área da medicina, engenharia de materiais, industriais, dentre outras. Da mesma forma que o campo elétrico de rastro produzido em um plasma pode ser utilizado para acelerar partículas de sorte a compor um feixe altamente energético, tal campo elétrico de rastro pode ser também utilizado para desacelerar os feixes, após sua utilização na aplicação pretendida. Desacelerar os feixes é importante pois partículas altamente energéticas causam a radioativação da estrutura, sendo os estágios desaceleradores também responsáveis em ocupar grande parte da máquina. Se o campo elétrico de rastro pode ser utilizado para acelerar as partículas carregadas às altas energias pretendidas em curtas distâncias, tal campo elétrico de rastro também pode ser utilizado para desacelerar os feixes altamente energéticos em também curtas distâncias, tornando as estruturas necessárias ao descarte do feixe extremamente diminutas. Nesses termos, o objetivo do presente trabalho é simular numericamente a desaceleração de feixes de partículas carregadas mediante o campo de rastro produzido. Os resultados obtidos serão comparados com resultados analíticos. |
|
Desenho de novos inibidores azólicos de corrosão de metais (chaer2sd) Universidade Federal do Rio de Janeiro Departamento de Físico-Química Coordenador: Marco Antonio Chaer do Nascimento Áreas do conhecimento: Ciência dos materiais; Química Início da vigência: 02-06-2017 A corrosão é um problema severo na indústria, responsável por perdas de bilhões de dólares por ano. Os gastos com corrosão nos Estados Unidos chegaram a cerca de 400 bilhões de dólares, o equivalente a 3 % do PIB do país, enquanto que no Brasil os gastos correspondem a 3,5 % do PIB. Em consequência disso, há uma grande atividade de pesquisa na área de corrosão, principalmente na busca por bons inibidores, já que este é o meio mais efetivo, econômico e flexível de controle da corrosão interna. Entretanto, a grande maioria das pesquisas nesta área é conduzida de forma empírica, na base da “tentativa e erro”, ou quando muito explorando relações do tipo estrutura do inibidor e sua eficiência. Esses procedimentos, além de muito caros e lentos, pouco contribuem para o entendimento da natureza do problema, ou seja, do mecanismo de corrosão, e consequentemente para o desenvolvimento de novos e mais eficientes inibidores de corrosão. Neste trabalho propomos uma abordagem teórico-experimental para entender o mecanismo de ação dos inibidores já conhecidos e projetar novos inibidores, mais eficientes e ambientalmente corretos. O mecanismo de adsorção será estudado através de cálculos quanto-mecânicos, periódicos e não periódicos, que nos permitirão entender de que forma o inibidor interage com a superfície metálica e o mecanismo de proteção da superfície. Esses estudos permitirão também determinar se há formação de filmes superficiais e as características desses filmes, dados importantes para a determinação do grau de recobrimento da superfície e, portanto, da concentração do inibidor a ser usada. As moléculas mais promissoras serão sintetizadas e testadas experimentalmente, quanto à sua eficiência de inibição, por meio de técnicas convencionais de perda de massa, medida de potencial de corrosão, e por técnicas eletroquímicas mais sofisticadas, tais como diagramas de impedância eletroquímica e microscopia eletroquímica de varredura. Os cálculos serão feitos em nível DFT, com condições periódicas de contorno, base de ondas planas e pseudopotenciais, utilizando o programa Quantum Espresso para realizar o estudo da interação das moléculas com as superfícies em diferentes graus de oxidação (Figura 4). Para cada potencial inibidor será feito um estudo sistemático para verificar como uma molécula se adsorve preferencialmente na superfície, como o processo de adsorção varia com o número de moléculas adsorvidas, o grau de recobrimento máximo que pode ser obtido com cada potencial inibidor e a distribuição das moléculas na superfície no regime de recobrimento máximo (formação de filme, empilhamento etc.) Essas informações guiarão as sínteses para obtenção e testes dos potenciais inibidores. Em cada caso, a estabilidade da camada protetora será testada adicionando-se algumas poucas moléculas de água ao sistema. As seguintes moléculas e seus derivados serão considerados no presente estudo: imidazol, imidazolina, imidazolidina e pirrolidina. O estudo da interação de uma única molécula de cada espécie com a superfície (001) de Fe já foi concluído. |
|
|
Desenvolvimento de funcionalidades, aumento da escalabilidade e estudos numéricos com modelos atmosféricos de alta resolução: BRAMS e OLAM (bramsolam) Universidade de São Paulo Instituto de Astronomia, Geofísica e Ciências Atmosféricas Coordenador: Pedro Leite da Silva Dias Áreas do conhecimento: Meteorologia Início da vigência: 28-06-2016 O modelo meteorológico regional utilizado nesse trabalho de equipe partiu do Regional Atmospheric Modeling System, inicialmente desenvolvido na Colorado State University, e passou por drásticas modificações, visando o paralelismo massivo, novas funcionalidades que melhor representam os fenômenos meteorológicos tropicais e acoplamento com a biosfera, oceanos, áreas inundadas, efeitos da emissão de aerossóis urbanos e por queimadas, química da atmosfera, com ênfase nos efeitos urbanos, emissões biogênicas etc. (Freitas et al. 2009). Somente como referência, foi o esforço brasileiro que elevou a escalabilidade da versão americana (RAMS) de algumas poucas centenas de núcleos para mais de 10.000 cores na versão brasileira denominada BRAMS – Brazilian Developments on the Regional Atmospheric Model (Souto et al. 2015). Mais recentemente, uma iniciativa nos EUA levou ao desenvolvimento de um modelo de domínio global, com núcleo dinâmico inovador, baseado em discretização por volumes finitos, e com toda a formulação dos processos físicos do RAMS/BRAMS, incluindo soluções numéricas e computacionais para garantir maior escalabilidade em plataformas computacionais de processamento massivamente paralelo. Esta versão é denominada OLAM – Ocean Land Atmosphere Model (Walko et al. 2008). Hoje, no Brasil, é feito um esforço integrado entre CPTEC/INPE, IAG/USP, LNCC, IME/USP, UFCG, UFSC e EMBRAPA de desenvolvimento do modelo e com uso para validação e operação na FUNCEME, UFRJ, UFSM, UFPA, INPA, UEA e vários centros regionais de Meteorologia através de um projeto de encomenda do MCTI. A proposta de uso do Santos Dumont no projeto BRAMS/OLAM tem os seguintes focos: 1. Paralelização: (a) OLAM - trabalho desenvolvido principalmente no LNCC com a participação do Dr. Jairo Panetta (ITA) que visa aumento de escalabilidade e migração de algumas partes do modelo para GPU e XEON-PHI e (b) BRAMS – trabalho desenvolvido principalmente pelo Dr. Jairo Panetta do ITA com parceira do CPTEC, LNCC e IAG, também visando o aumento da escalabilidade com foco nas aplicações operacionais do modelo. 2. Utilização do BRAMs/OLAM nas seguintes linhas de pesquisa: 2.1 Assimilação de dados de microfísica da precipitação no BRAMS 2.2 Acoplamento do BRAMS a um sistema de assimilação de dados convencionais e de satélite através do sistema GSI 2.3 Circulações locais na Amazônia e organização da precipitação com malhas de alta resolução no OLAM com ênfase no efeito das áreas alagadas: 2.4 Análise dos erros de previsões de tempo com alta resolução no BRAMS e OLAM do campo de vento para aplicações no gerenciamento de parques eólicos 2.6 Melhorias da funcionalidade do acoplamento com aerossóis e gases traço no BRAMS 2.7 Acoplamento do BRAMS com processos oceânicos 2.8 Efeito do acoplamento troposfera/estratosfera através do ozônio no OLAM. |
|
|
Desenvolvimento de Métodos Teóricos para o estudo da relação estrutura-funcionalidade em Biomoléculas (dynemol_ufsc) Universidade Federal de Santa Catarina Departamento de Física Coordenador: Luís Guilherme de Carvalho Rego Áreas do conhecimento: Ciência da computação; Física; Química Início da vigência: 17-12-2018 O objetivo geral deste projeto é estudar os mecanismos microscópicos responsáveis pelo transporte de carga e energia em biomoléculas e proteínas, bem como desenvolver métodos teórico/computacionais para elucidar o papel das coerências clássicas e quanticas na funcionalidade de sistemas biofísicos. Uma das perguntas centrais que pretendemos responder com essa pesquisa é: De que modo a energia de um fóton, ou de uma ligação química, é convertida em energia útil para o funcionamento de um organismo vivo? Em muitos casos, a absorção de um fóton desencadeia uma sequência de processes ultra-rápidos de transferência eletrônica, como ocorre nos primeiros estágios da fotossíntese, por exemplo. A energia potencial dos fragmentos oxidados ou reduzidos pela transferência eletrônica, por sua vez, induz transporte de ions e a formação de novos compostos. Tais processos de transferência eletrônica, em muitos casos, envolvem o movimento coordenado de elétrons e núcleos e o tunelamento eletrônico entre centros doadores e aceitadores localizados na proteína. O tunelamento eletrônico é facilitado pelo fenômeno de super-exchange (tunelamento através de estados virtuais) pois a proteína é essencialmente isolante. Um dos desafios centrais desse campo de pesquisa é entender como a estrutura da proteína, que circundeia os estados doador e aceitador, assim como sua dinâmica influenciam a transferência eletrônica de longo alcance nas proteínas. Dentre os problemas de interesse estão: (1) transferência de carga e energia em sistemas moleculares, (2) transdução fotoinduzida em sistemas moleculares, (3) efeitos quânticos em dinâmica molecular, (4) coleta e conversão de energia luminosa em sistemas naturais e sintéticos. O grupo Dinemol desenvolvemétodos híbridos do tipo QMMM (Quantum Mechanics/Molecular Mechanics) capazes de descrever efeitos de acoplamento elétron-núcleo em sistemas moleculares complexos. Os métodos computacionais desenvolvidos pelo grupo estão implementados no programa Dynemol, de livre acesso em https://github.com/lgcrego/Dynemol. O programa faz uso intenso de processamento paralelo através dos protocolos openMPI e MPI, além de GPU. |
|
|
Detecção de plágio multilíngue em artigos biomédicos (dpm) Universidade Federal do Rio Grande do Sul Engenharia de Produção Coordenador: Michel Jose Anzanello Áreas do conhecimento: Ciência da computação; Engenharias; Linguística, letras e artes Início da vigência: 21-06-2018 Plágio é a utilização do trabalho de uma terceira pessoa sem o devido reconhecimento ou citação, sendo um dos principais problemas na academia e na ciência. Este comportamento antiético está crescendo rapidamente, alimentado especialmente pela facilidade em compartilhar e recuperar informações através da internet. O desenvolvimento de soluções para detecção automática de plágio (PD) pode auxiliar humanos na identificação de conteúdos possivelmente copiados, além da sua fonte, mas também devem ser capazes de identificar casos de plágio multilíngue, o qual está se tornando recorrente, especialmente pelo fato de ferramentas de tradução automática atuais atingirem performance excepcional. Esta proposta de pesquisa vise desenvolver novas abordagens para detecção de plágio multilíngue em artigos biomédicos, dado que esta área científica em alto crescimento pode beneficiar-se de métodos que levem em consideração as características e terminologias não usualmente vistas em outras áreas científicas. Inicialmente, esta pesquisa irá focar no uso de ferramentas de tradução neural para derivar word embeddings, os quais serão utilizados para computar a similaridade semântica textual entre segmentos. O principal output no estágio inicial será a criação de modelos de tradução utilizando deep learning. Tais modelos tem cada vez mais se mostrado superiores aos tradicionais métodos estatísticos, porém requerem alto poder computacional. Atualmente não existe modelo de tradução já treinado para área biomédica, além dos tradutores disponíveis (ex: Google e Bing) não terem API gratuita. |
|
DockThor-VS: Web Server para Triagem Virtual de Fármacos em Larga Escala (dockthorvs) Laboratório Nacional de Computação Científica Coordenação de Mecânica Computacional Coordenador: Laurent Emmanuel Dardenne Áreas do conhecimento: Ciências biológicas Início da vigência: 17-08-2016 Técnicas computacionais, conhecidas como atracamento molecular (do inglês, molecular docking), que sejam capazes de prever o modo de ligação e os detalhes do tipo de reconhecimento molecular proteína-ligante, assumem cada vez mais um papel fundamental em áreas estratégicas de pesquisa associadas à saúde e à biotecnologia, que podem envolver centenas de milhões de dólares. Cada vez mais, indústrias farmacêuticas e grupos de pesquisa, que trabalham na busca de novas moléculas candidatas a fármacos, necessitam de metodologias mais rápidas, eficazes e de baixo custo. Neste cenário, a triagem virtual em larga escala baseada na estrutura tridimensional do receptor tem se destacado como uma importante ferramenta na busca de compostos protótipos. A triagem virtual baseada em estrutura consiste em analisar computacionalmente uma grande quantidade de ligantes com o objetivo de selecionar, de acordo com os modos de ligação e as afinidades de ligação previstos, compostos provavelmente mais ativos farmacologicamente para determinada doença. Nos últimos doze anos o Grupo de Modelagem Molecular de Sistemas Biológicos do Laboratório Nacional de Computação Científica (LNCC) tem desenvolvido o programa DockThor para predição de complexos moleculares receptor-ligante. Mais recentemente foi lançado em nível nacional, na 65a Reunião Anual da Sociedade Brasileira para o Progresso da Ciência (SBPC), o Portal web DockThor (www.dockthor.lncc.br), disponibilizando gratuitamente para a comunidade acadêmica o primeiro programa brasileiro (e possivelmente de todo o hemisfério sul) de atracamento molecular receptor-ligante. O portal está acoplado às facilidades de computação de alto-desempenho do SINAPAD. Desde o seu lançamento o portal DockThor foi acessado por mais de 3.500 visitantes únicos e teve cerca de 3500 jobs submetidos. Com a instalação do supercomputador brasileiro Santos Dumont abre-se uma oportunidade excepcional para o desenvolvimento de uma plataforma computacional, envolvendo metodologias e programas próprios, para triagem virtual em larga escala de compostos candidatos a fármacos que possa ser utilizada por grupos de pesquisa e empresas brasileiras atuantes nas áreas de química medicinal e biotecnologia. O presente projeto objetiva implementar a primeira etapa do desenvolvimento do portal web DockThor-VS (VS de virtual screening) para triagem virtual em larga escala acoplado ao Santos Dumont. A versão atual do portal DockThor permite apenas realizar experimentos computacionais envolvendo um ligante e um receptor por vez. No DockThor-VS será possível investigar bibliotecas de ligantes contendo milhões de moléculas contra dezenas ou até mesmo centenas de alvos moleculares. É importante ressaltar que pesquisadores ligados a este projeto são membros do INCT-INOFAR (Instituto Nacional de Ciência e Tecnologia de Fármacos e Medicamentos - http://www.inct-inofar.ccs.ufrj.br/) onde desenvolvem pesquisas aplicadas de desenho racional de fármacos através de fortes colaborações com grupos de pesquisa e instituições relevantes na área de desenvolvimento de novos fármacos para o tratamento de doenças negligenciadas, Alzheimer e inflamatórias crônicas. |
|
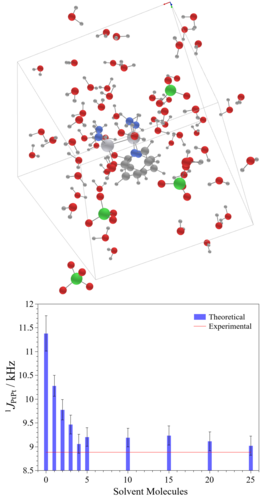
Efeito do solvente em complexos dinucleares de platina(III) via dinâmica molecular ab initio e cálculos de tensores blindagem e constante de acoplamento spin-spin (aimdsolv) Universidade de São Paulo Instituto de Química Coordenador: Lucas Colucci Ducati Áreas do conhecimento: Física; Química Início da vigência: 25-06-2018 Complexos de diplatina platina (Pt-Pt) e derivados vem sendo investigados para potenciais aplicações como drogas antitumorais devido às suas baixas associaçõescom nefrotoxicidade. Estes compostos apresentam relevâncias tecnológicas, como propriedades magnéticas, ópticas, quantum dots, bem como, processos industriais catalíticos. No entanto, a compreensão estrutural e espectroscópica destes compostos é ainda muito limitada em solução, o que carece de novas metodologias que forneçam melhores informações, principalmente em solução, onde grande parte dos complexos são sintetisados, acarretando melhor entendimento para futuras aplicações tecnológicas. A Ressonância Magnética Nuclear (RMN), a técnica mais poderosa para determinação estrutural em solução, tem em seus parâmetros tensor blindagem (σ)/deslocamento químico (δ), e as constantes de acoplamento (J) fontes de informação para determinação inequívoca da estrutura molecular, dependendetes da estrutura molecular e eletrônica que é fortemente perturbada pela sovatação. Para melhor elucidar a dinâmica da estrutura soluto solvente e como muda a densidade eletrônica dos complexos Pt-Pt, cálculos de dinâmica molecular ab initio e da teoria do funcional de densidade serão aplicados neste estudo, para simulação das espécies em solução. Estudos prévios com solvatação implícita anteriores sugerem que a descrição explícita do sistema soluto-solvente de forma dinâmica, possibilitará a descrição mais realista das condições experimentais, melhorando a precisão dos valores de σ, δ e J com geometrias estáticas em nível DFT, com inclusão de efeito de solvente implícito e Hamiltoniano com correção relativística, superestimam os valores experimentais em cerca de 100% para a maioria dos compostos. Para isso, a dinâmica molecular de Kohn-Sham (KS) Car-Parrinello (CPMD) e cálculos KS híbridos de propriedades magnéticas, com e sem efeito de solvente, serão utilizados. Este projeto propõe uma nova aplicação estado da arte no estudo dos parâmetros espectroscópicos e da dinâmica soluto solvente e estruturais em solução, o que auxiliará na elucidação do comportamento catalítico e outras propriedades em que o papel do solvente é fundamental. |
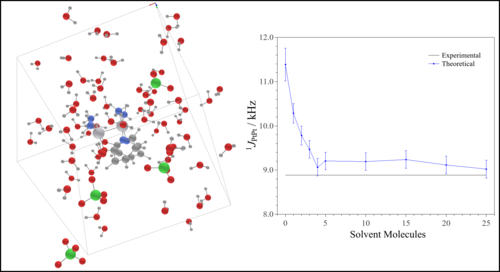
|
Efeito do Solvente em Parâmetros Espectroscópicos de RMN por Dinâmica Molecular ab initio (nmrmd) Universidade de São Paulo Instituto de Química Coordenador: Lucas Colucci Ducati Áreas do conhecimento: Física; Química Início da vigência: 03-08-2018 Ressonância Magnética Nuclear (RMN) é a técnica mais poderosa para determinação estrutural em solução. Os parâmetros como tensor blindagem (σ)/deslocamento químico (δ), e as constantes de acoplamento (J) são as fontes mais importantes de informação para determinação inequívoca da estrutura molecular e conformação adotada por uma molécula em solução. δ e J dependem da estrutura molecular e eletrônica, e serão suportados por valores calculados através da mecânica quântica. Nesta proposta são avaliados o efeito de sovatação da água e, principalmente, a contribuição da ligação de hidrogênio entre solvente-soluto no δ de 14N de compostos nitrogenados. Estudos teóricos usando modelos de solvatação implícita anteriores sugerem o uso de solvatação explícita e a descrição do sistema soluto-solvente de forma dinâmica, a fim de obter uma descrição mais realista das condições experimentais, melhorando a precisão dos valores estimados. Além disso, influência da coordenação do solvente (água) sobre o σ, δ e J de complexos Pt-Pt, precursores dos sais de Magnum, também será alvo de estudo. Esses complexos se encontram em equilíbrio em meio aquoso, sendo necessário o uso de sovatação explícita e uma descrição do sistema soluto-solvente de forma dinâmica. Os valores teóricos de σ, δ e J com geometrias estáticas em nível DFT, com inclusão de efeito de solvente implícito e Hamiltoniano com correção relativística, superestimam os valores experimentais em cerca de 100% para a maioria dos compostos. Para isso, a dinâmica molecular de Kohn-Sham (KS) Car-Parrinello (CPMD) e cálculos KS híbridos de propriedades magnéticas, com e sem efeito de solvente, serão utilizados. Esta nova técnica computacional, aliando KS-CPMD e cálculos KS-DFT híbridos para determinação de propriedades magnéticas, foi proposta e desenvolvida durante os últimos 2 anos durante o estágio de pesquisa deste proponente no exterior, e tendo sido aplicada com sucesso para compostos de metais de transição Tl-Pt. Este projeto propõe uma nova aplicação desse tipo de estudo de parâmetros espectroscópicos em solução, com o intuito de consolidar uma nova linha de pesquisa, inédita no país, bem como melhorar a formação em alto nível dos alunos. Há uma forte colaboração com grupos experimentais do IQ/USP e um do exterior. |
|
|
Efeitos isotópicos sobre as propriedades elétricas de isotopólogos moleculares: aplicações em astroquímica e nanociência (isotope) Centro Federal de Educação Tecnológica de Minas Gerais Coordenação de Área de Ciências Coordenador: Antônio Francisco Cruz Arapiraca Áreas do conhecimento: Astroquímica; Física; Química Início da vigência: 25-01-2019 Neste projeto pretendemos realizar um estudo teórico e computacional, utilizando cálculos ab initio e de teoria do funcional densidade, do efeito isotópico sobre propriedades elétricas de diversos sistemas de isotopólogos moleculares de larga escala como hidrocarbonetos aromáticos policíclicos (PHA), fulerenos funcionalizados e complexos endoedrais. O estudo destas propriedades será feito na perspectiva de aplicações nos contextos de astroquímica e nanociência. No caso astroquímico, por exemplo, intensidades de transições do espectro rotacional puro, de modo a prover informações que auxiliem observações via radiotelescópios, podem ser obtidas a partir dos momentos dipolares calculados para um conjunto de hidrocarbonetos aromáticos policícliclos deuterados (PAD) e fulerenos funcionalizados com hidrogênios e deutérios. Na seara da nanociência, além do efeito isotópico sobre os momentos dipolares de complexos de fulerenos endoedrais, pretendemos quantificar o efeito de blindagem devido à polarização dipolar que o sistema hospedeiro sofre devido à presença do sistema hóspede polar. Para tal estudo utilizaremos uma correção adiabática variacional de uso generalizado, que tem capacidade de computar os efeitos de massa nuclear finita que implicam em quebra de simetria por substituição isotópica, em conjunto com uma metodologia perturbativa que leva em conta correções vibracionais que consideram o campo de força anarmônico e os desvios da linearidade das propriedades moleculares. Essa metodologia já fio capaz de calcular, com boa acurácia na comparação com dados experimentais, os momentos dipolares para uma gama considerável de moléculas poliatômicas. |
|
|
Engenharia de Proteínas e Biomiméticos com Potencial em Diagnóstico e Vacinal para os Vírus Dengue e ZIKA (biomatdesign) Centro de Pesquisas Aggeu Magalhães, Fundação Oswaldo Cruz Virologia Coordenador: Roberto Dias Lins Neto Áreas do conhecimento: Ciências biológicas; Química Início da vigência: 10-10-2019 A detecção de antígenos e anticorpos específicos de patógenos é um dos métodos mais utilizados em diagnóstico clínico. Tal procedimento é utilizado no diagnóstico de câncer, doenças infecciosas, autoimunes, etc. Além disso, as moléculas de anticorpos constituem o mecanismo de proteção biosintética mais efetivo. Um aspecto crítico e até mesmo limitante para a síntese de anticorpos altamente específicos é o fato de que alguns epítopos podem se apresentar de forma transiente, expondo-se apenas quando o vírus se liga ao receptor. Uma alternativa para tal problema é a engenharia de estruturas moleculares (proteínas naturais ou biomiméticos) que possam servir de suporte para tais epítopos. De natureza multidisciplinar, métodos de simulação computacional serão sinergisticamente associados a técnicas experimentais de biologia molecular, imunologia e biofísica molecular para o desenvovimento de moléculas potencialmente imunorreativas. Especificamente, este projeto tem por objetivo o desenho racional e a caracterização das bases moleculares de proteínas e biomiméticos de interesse imunológico (diagnósticos e vacinais) que tem como alvo os vírus Dengue e ZIKA. O desenvolvimento dessas novas moléculas podem contribuir não apenas na área de saúde publica, mas também para a redução da nossa dependência tecnológica, a partir da produção de insumos com tecnologias estritamente nacionais. |

|
Ensino de Computação de Alto Desempenho - HPC - na graduação do curso de Sistemas de Informação (hpcfai) FAI - Centro de ensino Superior em Gestão, Tecnologia e Educação Departamento de Informática Coordenador: Fábio Gavião Avelino de Méllo Áreas do conhecimento: Ciência da computação; Sistemas de informação Início da vigência: 23-07-2019 O fator inovador do presente projeto consiste na disseminação da computação de Alto desempenho - HPC - para alunos de graduação em Sistemas de Informação e cursos afins. Espera-se com a iniciativa despertar o interesse de estudantes para esta área. O amadurecimento do ensino desta disciplina gerará novos projetos de Iniciação Científica, despertará o interesse de diversos cursos de extensão em replicar a disciplina nos mesmos e também em cursos de pós-graduação strictu sensu e lato sensu na região. É esperado também que haja a criação de projetos para empresas focadas em tecnologia que poderão ter interesse em usar HPC para seus projetos de Inteligência Artificial e Sistemas de Apoio a Decisão como, por exemplo, os que usam Otimização Combinatória. |
|
|
Estimação Não Invasiva de Reserva Fracionada de Fluxo Coronário FFR (hmlffr2) Laboratório Nacional de Computação Científica Coordenação de Métodos Matemáticos e Computacionais Coordenador: Pablo Javier Blanco Áreas do conhecimento: Ciências da saúde; Engenharias Início da vigência: 26-09-2019 O objetivo do presente projeto é dar continuidade às atividades de pesquisa e desenvolvimento sobre técnicas de modelagem computacional e simulação numérica na área de hemodinâmica computacional do sistema cardiovascular humano. Este projeto constitui a continuação do projeto do mesmo nome, que for a aprovado no ano 2017. Especificamente, o projeto visa o desenvolvimento de modelos baseados em dinâmica de fluidos computacional e modelos de dimensão reduzida para realizar o cálculo numérico da reserva fracionada de fluxo coronário. Isto é, desenvolver métodos numéricos para estimar índices fluidodinâmicos associados à presença de lesões de caráter obstrutivo em artérias coronárias, e a partir disto realizar a análise de risco de isquemia miocárdica. Na versão anterior do projeto foram desenvolvidos modelos orientados para pacientes específicos para simular a circulação na árvore coronariana podendo considerar presença de estenoses e o efeito de vasodilatadores, sendo que foram realizados diversos estudos, tais como o desenvolvimento de modelos 1D (ver produção) em detrimento do estudo do efeito de vasos colaterais. Nesta fase do projeto, procar-se-á validar, frente a modelos 3D completos, a capacidade de predição de modelos de compartimentos (modelos 0D) e modelos 1D enriquecidos (modelos 1D+). Como no projeto anterior, a anatomia vascular coronária de cada paciente será obtida a partir do processamento e segmentação de imagens médicas. Em segundo lugar, estes modelos geométricos serão empregados para estimar índices cardiovasculares de interesse médico utilizando diferentes modelos matemáticos, e avaliar o risco de isquemia em pacientes com doença coronária. Ainda, realizar-se-á um estudo com uma população de pacientes mais ampla, podendo chegar a superar a centena de casos de estudo. Espera-se que os resultados alcançados com este trabalho venham a fornecer ferramentas não invasivas, com grau de confiabilidade equivalente às técnicas invasivas, que auxiliem na avaliação de risco de isquemia de miocárdio em pacientes com doença coronária. |
|
|
Estrutura eletrônica e transporte em nanodispositivos de baixa dimensionalidade (nanosys) Universidade Estadual Paulista Júlio de Mesquita Filho Departamento de Engenharia Mecânica Coordenador: Ricardo Paupitz Barbosa dos Santos Áreas do conhecimento: Física Início da vigência: 02-01-2020 O presente projeto propõe o aprofundamento de estudos realizados a respeito de sistemas nanoscópicos, especialmente os bi-dimensionais, realizados nos últimos anos no laboratório de simulação computacional do departamento de Física da Unesp de Rio Claro. A proposta de uso dos recursos de computação de alto desempenho disponíveis no Santos Dumont visa permitir o cálculo de propriedades estruturais e eletrônicas de sistemas bidimensionais que têm potencial para a construção de novos nanodispositivos semicondutores baseados em novos materiais que o grupo vem estudando nos últimos 7 anos. O projeto conta com a colaboração científica do grupo do prof. Adri C. T. van Duin (Penn State University), criador do potencial ReaxFF e com a colaboração do Prof. Pekka Koskinen, da Universidade de Jyvaskyla (Finlândia). A contribuição do Prof. Koskinen estará, principalmente no desenvolvimento de novos parâmetros para a aproximação Tight Binding Density Functional Theory (DFTB) que utilizaremos de modo intenso durante o projeto. O DFTB, por ser uma aproximação do DFT, nos permite obter informações de caráter quântico sobre estruturas com grande número de átomos (centenas ou milhares, dependendo dos recursos computacionais disponíveis). Os recursos solicitados no presente projeto são essenciais para possibilitar que sejam realizadas buscas exaustívas para determinar as relações entre propriedades estruturais e propriedades eletrônicas dos materiais e dispositivos de interesse. Propomos o estudo de junções entre materiais semicondutores estruturalmente relacionados com o grafeno, como o Grafenileno (recentemente sintetizado por dois grupos experimentais diferentes)e versões modificadas, seja por dopagem seja por modificações geométricas a fim de determinar se seria possível controlar de algum modo o transporte eletrônico ou outros efeitos eletromecânicos, como a peizoeletricidade. Para a realização de tais estudos é necessário desenvolver novos parâmetros DFTB para os elementos químicos envolvidos e o uso dos recursos solicitados no presente projeto será determinante para o sucesso do estudo em tempo razoável. Como possíveis aplicações práticas temos a possibilidade de serem produzidos em laboratório, tendo em vista que parte dos materiais propostos já foram obtidos experimentalmente por diferentes grupos internacionais de pesquisa. |
|
Estudo da interação de peptídeos antimicrobianos (PAMs) com modelos de membrana celular por simulações de Dinâmica Molecular (antimicmd2) Universidade Estadual Paulista Júlio de Mesquita Filho Departamento de Física Coordenador: Alexandre Suman de Araujo Áreas do conhecimento: Ciência da computação; Ciências biológicas; Física Início da vigência: 28-06-2019 A descoberta de fármacos capazes de combater bactérias, os antibióticos, representou uma revolução na medicina, salvando milhões de vidas e contribuindo para um incremento significativo na qualidade e expectativa de vida do ser humano. Entretanto, o uso indiscriminado desse medicamento vem selecionando cepas bacterianas resistentes a um amplo espectro de antibióticos. Assim, o desenvolvimento de novos medicamentos que atuem por vias antibióticas ainda não exploradas e, principalmente, de difícil adaptação, é de grande interesse na atualidade. Os peptídeos antimicrobianos (PAM) são pequenas particulas proteicas produzidas pelo sistema imune de alguns animais e vegetais e que apresentam ação, além de bactericida, contra vírus, fungos e células cancerosas. Os PAMs, que são catiônicos em pH fisiológico, etuam sobre a membrana celular das bactérias, a qual é constituída de fosfolipídeos aniônicos, induzindo poros e vazamento do conteúdo celular (lise celular). Como a constituição da membrana celular é uma característica fundamental em todo ser vivo, mutações que resultem em resistências aos PAMs são raras e inviáveis, indicando que a via antibiótica dos PAMs tem grande potencial de exploração. O crescimento vertiginoso no poder computacional observado nas últimas décadas, tanto em nível de software como de hardware, permitiu a aplicação de técnicas de simulação computacional, antes restritas a sistemas simples e com poucos elementos, a sistemas biológicos complexos. Esse avanço colocou as simulações computacionais de sistemas biológicos em pé de igualdade com as mais tradicionais técnicas experimentais de investigação em biofísica, de modo que, atualmente, grande parte dos trabalhos nessa área utilizam técnicas experimentais e computacionais, colaborativamente, para descrever e entender os sistemas de interesse. Dentre a miríade de métodos de simulações computacionais existentes, o mais utilizado atualmente para o estudo de sistemas contendo moléculas biológicas é a Dinâmica Molecular (DM) e suas variantes. Esse método fornece resultados precisos e confiáveis, além de amostrar de forma satisfatória o enorme conjunto de conformações que as biomoléculas podem assumir, o que é exigência básica para o cálculo de propriedades macroscópicas a partir de informações do sistema microscópico. O fato das conformações amostradas pela DM estarem conectadas no tempo, na forma de uma trajetória, permite a obtenção de uma gama de propriedades do sistema que vão desde a conformação mais provável (de equilíbrio) até correlações temporais de propriedades coletivas. No presente projeto propomos o estudo do processo de adsorção e ação de PAMs naturais e sintéticos sobre bicamadas lipídicas, usadas como modelo de membrana celular, por meio de simulações de Dinâmica Molecular no equilíbrio, cálculos de energia livre e Dinâmica Molecular a pH Constante. A observação, em nível atômico/molecular, das interações peptídeo/bicamada e a conformação que assumem em diferentes situações físico-químicas podem nos fornecer indícios das propriedades fundamentais dos peptídeos que lhes conferem capacidade bactericida, o que é fundamental para o desenvolvimento de novos antibióticos mais potentes, eficazes e com baixa propensão e gerar cepas resistentes. |
|
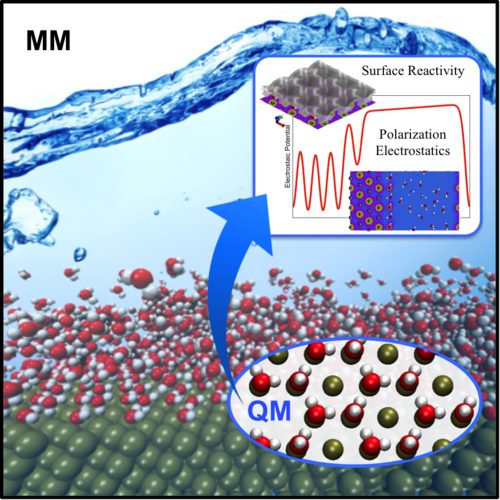
Estudo de Água em Interfaces Eletroquímicas (watredox) Universidade Federal do ABC Centro de Ciências Naturais e Humanas Coordenador: Mauricio Domingues Coutinho Neto Áreas do conhecimento: Ciência dos materiais; Física; Química Início da vigência: 16-07-2018 Interfaces eletroquímicas são componentes fundamentais em diversas aplicações tecnológicas. Desta maneira, o entendimento e controle dessas interfaces a nível microscópico é essencial para o desenvolvimento e consolidação de tecnologias como sistemas fotovoltáicos, sensores biológicos, etc. Particularmente, considerando que na maioria dos processos eletroquímicos, tais como eletrólise e eletrocatálise, tem-se água na forma de solvente ou reagente, é de extrema importância compreender os processos cinéticos e reações que ocorrem nas interfaces envolvendo água e eletrodos, bem como compreender as propriedades microscópicas da água. Assim, o uso de simulações atomísticas se torna fundamental para se obter uma compreensão mais detalhada em nível atômico e molecular. Considerando a natureza quântica dos fenômenos que ocorrem em interfaces eletroquímicas, simulações de primeiros princípios precisam ser consideradas. Nesta proposta propomos abordar duas questões de natureza fundamental para descrição de processos em eletrodos metálicos. No primeiro estudo propomos desenvolver e aplicar protocolos computacionais multiescala baseados em métodos QM/MM e cálculos de dinâmica molecular ao estudo de interfaces eletroquímicas envolvendo água e eletrodos metálicos de interesse para aplicações em energia e sensores biológicos. A motivação para o uso de métodos híbridos QM/MM e abordagens multiescala é a de contornar limitações de escala e tempo tipicamente presentes em simulações ab-initio. Simultaneamente pretendemos estudar as propriedades da água líquida e na interface com eletrodos metálicos utilizando técnicas de Machine Learning. Para aplicações em física, ML permite descobrir quais são as leis que produziram os dados usados no treinamento sem ter que assumir nenhuma forma específica para estas a priori, uma grande vantagem em relação aos processos de ajuste convencionais, nos quais se dá exatamente o oposto. Esta flexibilidade é importante no ajuste de funções (ou leis) complexas, como superfícies de energia potencial, pois permitem que a rede neural capte comportamentos sutis e difíceis de perceber, gerando assim, predições acuradas de forma computacionalmente mais eficiente. O objetivo desta etapa é o de desenvolver potenciais interatômicos (campos de força) para descrever as forças nas moléculas de água melhores do que os atualmente disponíveis na literatura. |
|
|
Estudo do acoplamento entre sawteeth and tearing modes no tokamak TCABR (ntm) Universidade de São Paulo Departamento de Física Aplicada Coordenador: Gustavo Paganini Canal Áreas do conhecimento: Física Início da vigência: 17-11-2017 The potential of nuclear fusion to provide a practically inexhaustible source of clean and renewable energy has motivated scientists to work toward developing nuclear fusion power plants. In this work, an outstanding issue encountered on the road toward the development of nuclear fusion will be addressed, namely the seeding of neoclassical tearing modes (NTMs) by sawteeth (ST) in tokamak plasmas. Although ST and NTMs have been intensively investigated over the last decades, a validated quantitative theory of the triggering of NTMs by ST remains to be developed. Many tokamaks observed that ST of sufficient duration may trigger NTMs that can lead to plasma performance degradation and/or disruptions, depending on the plasma conditions. From constraints imposed on the plasma performance, the onset of NTMs poses a threat to the main goal of ITER, which is a prototype intended to demonstrate sustained burning plasma operation with a fusion power gain factor Q = 10. This work seeks to investigate the mechanisms of NTM seeding by ST using the TCABR tokamak, located at the Laboratório de Física de Plasmas - LFP, Instituto de Física, Universidade de São Paulo. Experiments will be carried out in order to investigate the non-linear dynamics involved in the NTM seeding by the modes generated during the ST crash. To provide a better understanding of the dynamics behind mode coupling, the experimental measurements will be interpreted by numerical modeling using M3D-C1, which is a state-of-the-art code for plasma modeling that has recently been developed at the Princeton Plasma Physics Laboratory, Princeton, USA. Understanding the different possible mechanisms behind NTM seeding is of utmost importance for the development of controlled thermonuclear fusion as an economically viable energy source. The knowledge acquired during this work could lead to the development of a model in which the relative contribution from distinct seeding mechanisms and their scalings are known. This model could ultimately then be used predictively to indicate zones of safer operation at higher plasma pressures in existing tokamaks worldwide and in the design of future devices. |
|
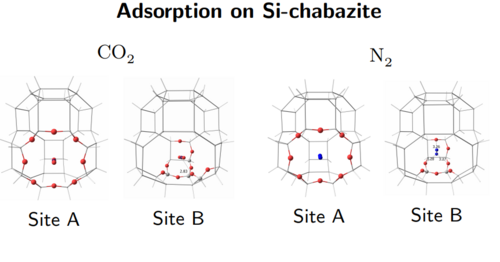
Estudo teórico-computacional de zeólitas com aplicações em catálise e adsorção (chaersd2) Universidade Federal do Rio de Janeiro Departamento de Físico-Química Coordenador: Marco Antonio Chaer do Nascimento Áreas do conhecimento: Ciência dos materiais; Química Início da vigência: 20-12-2019 Trata-se da renovação de um projeto já apoiado pelo LNCC que é, na verdade, um sub-projeto do Núcleo de Excelência em Funcionalização de Hidrocarbonetos, que visa desenvolver métodos para a produção de compostos químicos, de alto valor, a partir de matéria prima abundante no país, através de reações de funcionalização de hidrocarbonetos, usando zeólitas. Em termos de aplicações práticas, este sub-projeto pretende ampliar a compreensão do mecanismo das reações catalisadas por zeólitas, e de sua ação adsorvente em processos industriais de grande importância econômica. A partir desta compreensão, será possível contribuir para o desenvolvimento de: (a) novos catalisadores para importantes processos industriais, sobretudo nas indústrias de refino de petróleo e petroquímica, capazes de promover reduções de custos e melhor aproveitamento do petróleo e do gas natural; (b) novos adsorventes para separações gasosas de interesse da indústria de petróleo e do gas natural, e da indústria de produção de gases especiais; (c) novos adsorventes para captura de dióxido de carbono emitido por grande fontes estacionárias, com efeitos positivos na redução do aquecimento global. |
|
|
High-Fidelity Numerical Simulations of Wall-Bounded Turbulent Flows (hfwbtf) Instituto de Aeronáutica e Espaço Divisão de Aerodinâmica Coordenador: João Luiz Filgueiras de Azevedo Áreas do conhecimento: Engenharias Início da vigência: 24-07-2018 A presente proposta foi criada a partir de um projeto de colaboração internacional entre o Instituto de Aeronáutica e Espaço (IAE), Universidade de Campinas (UNICAMP), Instituto Tecnológico de Aeronáutica (ITA) e École Nationale Supérieure d'Arts et Métiers (ENSAM-ParisTECH), da França. A mesma visa diversas aplicações científicas e tecnológicas, como por exemplo, na área de energia eólica, engenharias mecânica e aeronáutica. O foco principal é o estudo de escoamentos compressíveis na presença de paredes sólidas, o que é típico em aplicações de engenharia e muito desafiador. As configurações investigadas incluem aerofólios, canais e trens de pouso. Existe o interesse em análise da física da turbulência, um dos "Millennium Prize Problems" de matemática, além da geração de ruído, tema atual devido às recentes restrições de emissão de ruído para aviação civil. Simulações de grandes escaladas, LES, com alta resolução e formulações híbridas RANS-LES, são as abordagens escolhidas para serem aplicadas na presente proposta. Estas abordagens serão aplicadas para os escoamentos de interesse com o objetivo de capturar mecanismos aerodinâmicos transientes e os correspondentes efeitos acústicos. Para o caso de estudo de escoamento sobre aerofólios e escoamentos em canais, esquemas numéricos de diferenças finitas de alta ordem e alta resolução serão empregados usando LES para a solução de estruturas turbulentas mais energéticas com o mínimo possível de adição numérica de fenômenos dissipativos e dispersivos sobre estruturas turbulentas. Com intuito de obter soluções acuradas, malhas estruturadas serão desenvolvidas para a resolução de camadas limites sobre as configurações de aerofólio e canais. Para o estudo de trens de pouso, a abordagem híbrida pode ser empregada para o cálculo dos escoamentos turbulentos. Este tipo de abordagem é capaz de capturar as estruturas mais energéticas do escoamento que são responsáveis pela geração de ruído. Esta formulação implica modelamento de turbulência sobre regiões onde as escalas de alta frequência estão presentes ou em regiões onde existe a presença de malhas de menor resolução. É importante lembrar que este tipo de abordagem é menos custosa quando comparada à formulação LES com resolução de parede. Diferentemente das configurações de aerofólio, trens de pouso apresentam diversos detalhes geométricos adicionais com diferentes escalas de comprimento. Esses detalhes não podem ser devidamente resolvidos usando malhas estruturadas e métodos de alta-ordem de precisão. Neste sentido, técnicas de discretização do tipo volumes finitos serão empregadas juntamente com malhas não estruturadas híbridas. |
|
|
Implementação e validação de um modelo de escoamento bifásico líquido-gás com diferentes escalas de interface (mflow) Universidade Federal de Santa Catarina Departamento de Engenharia Mecânica Coordenador: Emilio Ernesto Paladino Áreas do conhecimento: Engenharias Início da vigência: 07-06-2019
Escoamentos bifásicos líquido-gás possuem papel importante em processos naturais e industriais, principalmente na indústria química e petrolífera. Escoamentos desse tipo abrangem inúmeras áreas e processos de engenharia, como por exemplo o projeto de equipamentos para transporte de fluidos, produção de petróleo e gás, sistemas de arrefecimento, etc. Em geral, escoamentos bifásicos líquido-gás podem apresentar diferentes padrões, afetando de modo complexo a sua hidrodinâmica e também a transferência de calor e de massa.\nDevido à complexidade dos diferentes padrões de escoamento presentes em situações reais, um grande esforço é dedicado a melhor compreensão de escoamentos bifásicos líquido-gás. Nos últimos anos, a fluidodinâmica computacional (Computational Fluid Dynamics, CFD em inglês) tornou-se uma ferramenta importante, permitindo uma análise detalhada dos mecanismos físicos que regem o fenômeno. Desse modo, o trabalho descrito aqui possui caráter fundamental e pode encontrar aplicações em sistemas multifásicos, como sistemas de medição, processos envolvendo transferência de calor e massa, sistemas de alta eficiência energética e demais.\n |
|
|
Inteligência Computacional em Processamento de Alto Desempenho (hubdigital_coppe) Universidade Federal do Rio de Janeiro Programa de Engenharia Civil Coordenador: Alexandre Gonçalves Evsukoff Áreas do conhecimento: Ciência da computação; Engenharias; Geociências; Meteorologia Início da vigência: 03-07-2019
Nos últimos anos, o aumento do volume de dados observados ou produzidos por simulação numérica e a capacidade de processamento dos computadores permitiram o desenvolvimento de novos algoritmos e modelos de redes neurais cada vez mais complexas, com milhares de unidades de processamento e milhões de parâmetros. Esses modelos são chamados de redes neurais profundas e são ajustados por algoritmos de aprendizado profundo (deep learning). Resultados recentes em problemas complexos como visão computacional e processamento de linguagem natural mostram que redes neurais profundas são capazes de realizar tarefas complexas de sistemas de inteligência computacional. Em problemas da ciência da natureza, modelos de redes neurais realizam a modelagem de relações espaciais de longo alcance em várias escalas de tempo (https://www.nature.com/articles/s41586-019-0912-1) |
|
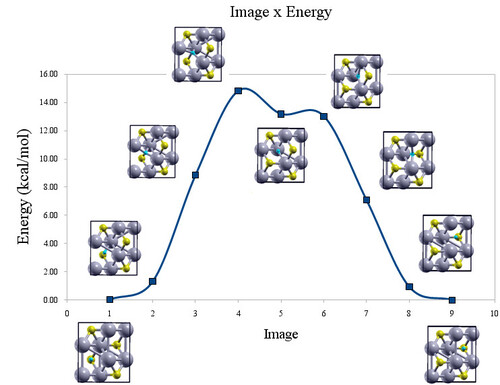
Mecanismo de oxidação de carbetos de metais de transição (moc) Universidade Federal do Rio de Janeiro Departamento de Físico-Química Coordenador: Alexandre Braga da Rocha Áreas do conhecimento: Ciência da computação; Engenharias; Química Início da vigência: 20-06-2018 Dentre as diversas aplicações de carbetos de metais de transição, destaca-se seu uso como catalisador em diversos tipos de reações, podendo substituir catalisadores mais caros, baseados em metais do grupo da platina. Os mais promissores carbetos são os de molibdênio, tungstênio, nióbio e vanádio. Esses já apresentam aplicações, não somente em catálise heterogênea como também na composição de baterias e capacitores. O controle da oxidação é um problema recorrente na química de carbetos, uma vez que a eficiência e seletividade destes como catalisadores depende do grau de oxidação da superfície. Na síntese desses materiais é importante o controle da quantidade de oxigênio na superfície exposta, onde a reação irá ocorrer. além disso, para trasnporte ou caracterização destes materiais, é costume estabelecer uma camada passivadora de óxido. No entanto, a eficiência desse processo é questionável devido à possibilidade de difusão de oxigênio nesta camada, destruindo ou modificando o material subjacente, que quer se proteger. Mecanismos de oxidação de superfícies envolvem não apenas a etapa de aumento da cobertura e de quebra da ligação O-O como também o estudo da difusão de oxigênio. Dessa foprma, cada uma dessas etapas deve ser investigada para um melhor controle do processo de oxidação desses materiais. Apesar da importância da oxidação na química de carbetos, existem poucos estudos na literatura sobre os mecanismos de oxidação. O estudo teórico das etapas de adsorção e difusão de oxigênio ajudam a elucidar este mecanismo em nível molecular. A análise das etapas do mecanismop de oxidação em nível teórico é complementar aos estudos feitos em laboratório e permite um entendimento do processo de oxidação, em nível molecular, que a maioria dos experimentos não fornece. Os resultados obtidos por cálculos ab initio podem auxiliar os experimentalistas no desenvolvimento de estratégias de controle na síntese e na conservação dos carbetos. Assim, carbetos podem ser projetados com propriedades otimizadas para umas aplicação desejada. Dessa forma, o objetivo do projeto é estudar, utilizando a teoria do funcional da densidade, os processos de adsorção e difusão de oxigênio nos referidos carbetos. O projeto visa a obtenção da energia de ativação para o processo de difusão de oxigênio, o coeficiente de difusão e o grau de cobertura de oxigênio. |
|
|
Métodos Quânticos e Empiricos Aplicados no Estudo e Desenvolvimento de Materiais para Aplicações em Novas Energias (qtnano2) Universidade de São Paulo Instituto de Química de São Carlos Coordenador: Juarez Lopes Ferreira da Silva Áreas do conhecimento: Física; Química Início da vigência: 09-04-2019
Estudos têm indicado que a contribuição da energia solar obtida através da conversão de fótons em elétrons via dispositivos semicondutores deve aumentar sua contribuição na matriz energética mundial de 1% para 30%, o que deve contribuir significativamente para para a redução da emissão de CO2 e, consequentemente, desaceleração do aquecimento global. O Brasil, como signatário das principais convenções internacionais sobre meio ambiente, deve adotar medidas de incentivo ao maior aproveitamento da energia solar, o que, entretanto, requer avanços científicos consideráveis, principalmente, no desenvolvimento de materiais para a construção de dispositivos de conversão energética com alta eficiência e baixo custo. Neste projeto, portanto, concentraremos nossos esforços em três classes promissoras de materiais, especificamente, calcogenetos, perovskitas, e materiais orgânicos, os quais foram selecionados devido às seguintes razões: |
|
|
Modelação e estatísticas de extremos de ventos e ondas no Oceano Atlântico (exwav) Universidade de São Paulo Centro de Engenharia e Tecnologia Naval e Oceânica Coordenador: Ricardo Martins Campos Áreas do conhecimento: Engenharias; Meteorologia; Probabilidade e estatística Início da vigência: 29-03-2019
A reconstituição da atmosfera e da agitação marítima utilizando modelagem numérica computacional (hindcasts) é extremamente importante para cálculos de parâmetros meteo-oceanográficos utilizados pela indústria offshore bem como para a segurança costeira. Apesar do aumento recente da cobertura e duração das medições com boias e satélites, os hindcasts ainda são a principal fonte de dados para o cálculo de valores extremos de longo período. Isso se deve à grande cobertura espacial e temporal, com informações contínuas e sem spikes. A importância e dependência dos hindcasts se tornam ainda maiores em regiões com poucas medições, como no Atlântico Sul. Dois pontos são importantes na construção dos hindcasts: 1) O sistema de assimilação de dados dos modelos atmosféricos aproximam as simulações para as condições reais presentes através das medições. Portanto, em regiões com baixa densidade de observações, os hindcasts têm erros muito maiores. 2) Os modelos numéricos são calibrados alterando os parâmetros internos a fim de minimizar certas métricas de erros, considerando todo espaço amostral (eventos calmos e extremos) e toda a cobertura espacial (geralmente o globo todo). Portanto, o impacto da qualidade dos eventos extremos no erro médio integrado é muito pequeno – justamente por serem raros e possuírem cobertura espacial limitada. Por esses motivos, os hindcasts costumam subestimar consideravelmente os eventos extremos e superestimar condições calmas. Isso gera dois problemas graves quando usados para ajustar distribuições de extremos: 1) A subestimação dos extremos é amplificada ao se extrapolar a função quantil, para o cálculo dos valores de retorno; 2) A subestimação dos extremos e superestimação das condições calmas têm um impacto nos momentos probabilísticos e nas distribuições, que são a base para os métodos tradicionais de cálculo de valores extremos. |
|
|
Modelagem Computacional de Reservatórios de Petróleo com Complexidade Geológica (simulreserv) Laboratório Nacional de Computação Científica Coordenação de Modelagem Computacional Coordenador: Marcio Rentes Borges Áreas do conhecimento: Ciências humanas; Engenharias; Matemática Início da vigência: 22-03-2018 Os reservatórios presentes na chamada camada pré-sal, localizados na costa brasileira, entre os estados do Espírito Santo e Santa Catarina, em uma faixa que se estende por cerca de 800km, representam um grande potencial petrolífero para o país. Tais reservatórios estão localizados a grandes profundidades, sob uma espessa camada de rochas salinas (com propriedades viscoelásticas), o que torna sua exploração um desafio científico e tecnológico. É neste contexto que, dentre os objetivos estratégicos previstos no Plano Diretor do LNCC, está a linha de pesquisa aplicada na área de Petróleo, Água e Gás que tem por objetivo consolidar a atuação do LNCC como um centro de referência nas áreas de modelagem numérica de processos de prospecção de óleo, gás natural e águas subterrâneas, bem como transporte de poluentes em solos e armazenamento de CO2 em reservatórios de petróleo e aquíferos, desenvolvendo pesquisa aplicada e formação qualificada de recursos humanos, com potencial de iteração com o setor produtivo. Em virtude disso, o LNCC faz parte do Programa de Recursos Humanos da Agência Nacional do Petróleo (PRH - 50 - Modelagem Computacional Hidro-Geomecânica de Reservatórios não Convencionais) que tem como objetivos: i) formar recursos humanos altamente qualificados para atender a demanda do setor de Petróleo e Gás; ii) ampliar as atividades de pesquisa do LNCC em áreas estratégias; iii)desenvolver novas formulações e metodologias computacionais orientadas para simulação de processos avançados de recuperação de petróleo e gás em reservatórios não convencionais; iv) atender à crescente demanda de candidatos aos programas de Mestrado e Doutorado em Modelagem Computacional do LNCC. Atualmente, a equipe de Simulação de Reservatórios do LNCC (da qual o proponente faz parte), no âmbito do projeto “Modelagem e Simulação Numérica de Escoamentos em Reservatórios Heterogêneos com Acoplamento Geomecânico” da rede SIGER (patrocinado pela PETROBRAS), vem desenvolvendo diversos códigos computacionais capazes de simular o escoamento de fluidos em reservatórios de petróleo heterogêneos, caracterizados pelo acoplamento hidro-geomecânico, fazendo uso de métodos numéricos localmente conservativos. Tais reservatórios, geralmente, são constituídos de rochas carbonáticas que podem apresentar estruturas geológicas complexas, tais como cavernas, vugs, juntas, falhas e veios, com uma vasta gama de heterogeneidades físicas e químicas que influenciam fortemente a produção de hidrocarbonetos. Cabe ressaltar que, a simulação numérica de problemas reais, postos em escalas de campo (da ordem de quilômetros), e discretizados na escala de laboratório (da ordem de centímetros e na qual as equações e as propriedades das rochas são conhecidas), dá origem a problemas computacionais de grande porte que exigem computação de alto-desempenho para que os mesmos sejam factíveis em tempo razoável. Então, o objetivo central do presente projeto é utilizar o potencial do supercomputador Santos Dumont para resolver problemas cada vez mais complexos e detalhados na área de simulação de reservatórios de petróleo, ajudando a indústria a tomar decisões acertadas com base nos conhecimentos gerados. |
|
Modelagem da Circulação no Oceano Atlântico Sul (modoceano2) Universidade Federal do Rio de Janeiro Programa de Engenharia Oceânica Coordenador: Afonso de Moraes Paiva Áreas do conhecimento: Engenharias; Geociências Início da vigência: 12-08-2019
O Projeto MODOCEANO é um projeto institucional do nosso laboratório na UFRJ, que visa o desenvolvimento de modelagem computacional, em alta resolução, da circulação oceânica em diferentes escalas espaciais e temporais no Atlântico Sul. São investigados aspectos relacionados: |

|
Modelagem da turbulência em escoamentos multifásicos de biorreatores com membranas (BRM) Universidade Federal do Rio Grande do Sul Departamento de Engenharia Química Coordenador: Nilo Sérgio Medeiros Cardozo Áreas do conhecimento: Engenharias Início da vigência: 26-05-2017 Os biorreatores com membranas (BRMs) possuem vantagens em relação ao tratamento convencional de efluentes, como maior eficiência e uma significativa redução de espaço físico necessário para o processo. O mercado de BRMs está consolidado em inúmeros países e tem apresentado altas taxas de crescimento. No Brasil, o crescimento da demanda por tratamentos adequados para os efluentes gerados na indústria têm incentivado a preferência pelos BRMs. Um problema conhecido e persistente no uso de BRMs é a ocorrência de incrustações nas membranas (fouling), que reduzem a eficiência do processo. O mecanismo de aeração promove a diminuição do fouling através do cisalhamento exercido pelas bolhas de ar sobre as membranas. Apesar da importância da hidrodinâmica multifásica em BRMs ser reconhecida por mais de vinte anos, as indústrias e a comunidade científica ainda não compreendem completamente os efeitos cisalhantes induzidos pelas bolhas de ar nos BRMs. O custo operacional associado à aeração é um problema recorrentemente mencionado; a aeração consome até 70% do total da energia de BRMs e é utilizada de forma otimizada apenas 10% das vezes. Apesar do crescente número de publicações sobre biorreatores com membranas, das quais mais de 25% trata de fouling, somente 4,3% estuda a hidrodinâmica nesses sistemas. Além disso, é crescente o número de publicações sobre BRMs no Brasil, mas não há nenhuma publicação brasileira relacionada à hidrodinâmica nesses sistemas. É evidente a necessidade do desenvolvimento de pesquisa e de tecnologia relacionadas à hidrodinâmica em BRMs no Brasil. Esse tipo de estudo normalmente emprega ferramentas de fluidodinâmica computacional (CFD), que resolvem numericamente problemas de escoamento de fluidos. Um dos primeiros passos para encontrar uma solução apropriada para simulações numéricas é propor uma modelagem correta. Dentre os fenômenos a serem modelados, a turbulência provavelmente é um dos mais importantes; contudo, são escassos ou inexistentes trabalhos que verifiquem a performance e a confiabilidade de modelos de turbulência na hidrodinâmica de BRMs. Além disso, para BRMs de grande porte como os utilizados em indústrias, normalmente são utilizadas funções de parede para a modelagem da camada limite; uma aproximação considerada inadequada quando se deseja obter valores de tensão cisalhante nas membranas, já que o cálculo dessa variável sofreria influência direta das aproximações realizadas através de funções de parede. Dessa forma, o objetivo desse projeto é verificar a importância da escolha do modelo de turbulência para simulações de biorreatores com membranas, com o uso de CFD, através do emprego de diferentes modelos de turbulência (k-ϵ e k-ω SST) e funções de parede (padrão, modificada e automática) e comparar os resultados da região da camada limite com modelos considerados mais adequados para tal (como k-ω SST sem função de parede; modelos do tipo LES, large eddies simulation; e DNS, direct numerical simulation) juntamente com dados experimentais a serem obtidos através da técnica PIV (particle image velocimetry) e dados provenientes da literatura. Espera-se que as conclusões obtidas venham a servir como base para desenvolver uma modelagem adequada desses sistemas, visando a otimização da intensidade de aeração e economia energética. |
|
|
Modelagem da Turbulência póximo à interface em escoamentos estratificados gás-líquido (nitcfd) Universidade Federal Rural do Semi-Árido Departamento de engenharia e Tecnologia Coordenador: Victor Wagner Freire de Azevedo Áreas do conhecimento: Engenharias Início da vigência: 15-08-2019
O presente projeto visa a simulação numérica direta (DNS) de um escoamento turbulento no padrão estratificado, levando em consideração a deformação da interface no regime de ondas capilares. O estudo da turbulência, de maneira geral, é sempre inovador porque, apesar dos trabalhos já publicados, o caráter estatístico dela sempre traz novas observações sobre o fenômeno, dependendo da abordagem utilizada. A escolha da DNS, apesar de computacionalmente mais cara, foi a melhor solução para podermos tirar as conclusões necessárias para o que está sendo observado. |
|
|
Modelagem de Materiais Funcionais (mmfufscar2) Universidade Federal de São Carlos Departamento de Química Coordenador: Andre Farias de Moura Áreas do conhecimento: Ciência dos materiais; Física; Modelagem Computacional; Química Início da vigência: 14-08-2019 Esta proposta de pesquisa pretende ser uma continuação do nosso projeto em andamento, que tem sido executado continuamente desde junho/2016, usando uma alocação premium no supercomputador SDumont. O foco geral do projeto ainda será o desenvolvimento computacional-experimental de materiais funcionais, embora nos últimos três anos tenhamos estreitado o escopo para nanomateriais quirais e mais recentemente para materiais também contendo múltiplos elétrons desemparelhados (e, portanto, tendo propriedades magnéticas intrínsecas). Como as vemos, estas não são apenas propriedades extras sendo medidas ou calculadas, mas produzem materiais com níveis sem precedentes de ajuste reversível, uma vez que as propriedades do materialtornam-se responsivas a campos elétricos e/ou magnéticos externos sem qualquer necessidade de executar modificações químicas irreversíveis. Além de melhorar a gama de aplicabilidade de qualquer material, renderizar nanoestruturas quirais também é um enorme salto em direção à biocompatibilidade de materiais e seu uso em aplicações médicas. Já realizamos uma série de investigações nessas novas direções e os resultados que obtivemos até agora mostram que estamos no caminho certo (os artigos estão listados na seção que descreve a produção científica associada à proposta). Deve-se ter em mente que a busca dessas novas fronteiras representa desafios para a modelagem computacional, bem como para a contraparte experimental. Do ponto de vista computacional, uma única investigação de uma nanopartícula modelo que é tanto quiral quanto magnética pode facilmente atingir um milhão de horas de CPU em apenas 1-2 meses. Apesar de exigentes, essas investigações têm sido compensadoras nos últimos três anos, conforme demonstrado pelos artigos publicados em periódicos de alto impacto (Science, Nature Chemistry, Chamical Communications, etc.), com uma expectativa realista de que continuaremos a publicar nessa faixa, desde que possamos manter nossa alocação premium funcionando. E o mais importante, manteremos as colaborações internacionais com os grupos do Prof. Nicholas Kotov (University of Michigan, EUA) e do Prof. Chuanlai Xu (Jiangnan University, PRC), cujas contribuições na contraparte experimental não podem ser superestimadas, juntamente com outras colaborações em diferentes estágios de maturação com pesquisadores de universidades e empresas locais. |

|
Modelagem Estocástica para Inversão Sísmica (meis2) Universidade Federal do Rio de Janeiro Programa de Engenharia de Sistemas e Computação Coordenador: Claudio Amorim Áreas do conhecimento: Ciência da computação; Geociências Início da vigência: 02-10-2018 Na indústria de Óleo e Gás, novas descobertas de reservas de petróleo são impulsionadas pelos avanços da tecnologia da exploração sísmica que depende da construção de modelos do subsolo cada vez mais precisos e fiéis a realidade. Neste projeto, as propriedades físicas das rochas no subsolo relacionadas à presença de hidrocarbonetos, tais como velocidade ou densidade, serão modeladas explorando a equação de propagação da onda sísmica através da técnica Full Waveform Inversion (FWI). O FWI é essencialmente um método de casamento dos dados adquiridos em campo com os dados produzidos pela modelagem, onde iterativamente tenta-se ajustar os parâmetros da equação de onda de tal forma que o resultado produzido pela modelagem coincida com os dados de campo. Trata-se, portanto, de um problema de otimização onde se busca encontrar o mínimo global da seguinte função de custo: M(m) = ||Dataacq-Datamod||2 Onde M(m) é o misfit, que representa a diferença entre os dados adquiridos (Dataacq) e os dados modelados (Datamod). Uma forma de estimar os parâmetros da modelagem é utilizar métodosde gradiente descendente, que embora sejam eficientes computacionalmente, podem ficar presos em mínimos locais, impedindo-os de atingirem o mínimo global. Uma forma de mitigar este problema consiste em oferecer um parâmetro inicial adequado de tal modo que, a partir deste ponto inicial, o método de gradiente seja capaz de convergir para a solução global. Uma forma de obter este valor inicial é utilizando Métodos de Monte Carlo, cuja abordagem probabilística evita que os mesmos fiquem presos em mínimos locais, no entanto, tais métodos possuem um custo computacional muito alto, pois a execução da modelagem pode alcançar um número de iterações muito elevado, da ordem de grandeza de 105 iterações. Como uma estratégia de solução do problema descrito, este projeto de P&D realizou as seguintes atividades: 1. Revisão bibliográfica para avaliar dentre as técnicas de Monte Carlo qual é a mais adequada ao problema de inversão sísmica que pretendemos abordar. Concluindo-se que o método de Metropolis-Hastings com Simulated Annealing constitui uma boa alternativa. 2. Foi selecionado na literatura um software para a geraçãodas amostras da distribuição prior utilizada pelo Metropolis-Hastings. O mesmo encontra-se em fase final de testes. 3. Foi desenvolvida e testada uma abordagem para o cálculo da função de custo de cada proposta gerada pelo simulador mencionado anteriormente. 4. Foi gerado um conjunto de dados sintéticos de teste para validar as técnicas desenvolvidas. 5. O workflow do Metropolis-Hastings foi implementado e encontra-se em fase de testes com os dados mencionados no item 4. As próximas etapas da pesquisa incluem: 1. Finalizar os ajustes no gerador de propostas prior assim como o workflow do Metropolis-Hastings. 2. Avaliar as demandas computacionais necessárias para o workflow estocástico, e propor novos mecanismos e técnicas que permitam tais aplicações se beneficiarem da próxima geração de supercomputadores exascale com início previsto para 2024. |
|
|
Molecular Mechanisms and interaction invloved on Dopamine Transport on Metabolism at the Nerve Terminals: Dopamine Transporter (DAT), a-Synuclein and Kinesin (datsk) Universidade Federal do ABC Centro de Matemática, Computação e Cognição Coordenador: Ana Ligia Scott Áreas do conhecimento: Ciências biológicas Início da vigência: 05-09-2019 A better comprehension of the molecular mechanisms that take part in several neurodegenerative illnesses (such as Alzheimer and Parkinsons disease, lateral amyotrophic sclerosis, and so on) can aid in the development of new drugs and treatments. One of the main pathologic characteristics present in all of the previously cited disorders is the present of insoluble protein aggregates. For example: Lewy bodies associates with Parkinsons and Lewy bodies dementia. This aggregated are made up mainly by the α-synuclein protein which presents a variety of conformations: monomeric and unstructured in physiological solution, multimeric (usually helical tetrameric), and cross-β-sheet-rich architecture of aggregates. The molecular mechanisms and interactions among Dopamine Transporter (DAT), α-Synuclein and Kinesin play an important role on Dopamine Metabolism at the Nerve Terminals. This research project aims to investigate molecular mechanisms involved in on Metabolism at the Nerve Terminals. It is intended to approach the target systems regarding several aspects: structure, structural dynamics (conformational changes/functional movements); α-synuclein aggregate formation and molecular interactions among: Dopamine Transporter (DAT), α-Synuclein and Kinesi (Brady and Morfini, 2017). We intend investigate molecular mechanisms and interactions of these macromolecules from different viewpoints and using a partial multi-scale modeling of the target systems system. This approach will allow for a better understanding of the molecular processes involved on metabolism at the Nerve Terminals that is important for some neurodegenerative diseases as Parkisnsons. Over the last three years, Dra. Ana Ligia Scott and her group has been financed by Fapesp to investigate molecules related to neurodegeneration and aggregate formation by means of normal modes analysis and molecular dynamics combination. The LBCB (our laboratory) has become more and more specialized in the study of macromolecules structural dynamics (functional movements), in collaboration with Dr. David Parahia and Dr. Maria Miteva, both from France and Ivet Bahar from MMBios - University of Pittsburgh. In 2017, Ana Ligia Scott was Professor Invite at Laboratoire de Biologies et Pharmacologie Appliquée (LBPA), Ecole Normale Supérieure de Paris-Saclay to study aggregation process. In 2018, The researcher took part in a sabbatical internship (post doctorate), financed by University of Pittsburgh and Fapesp to study molecular mechanism of Neurodegenerative Diseases at The National Center of Multiscale Modeling of Biological Systems (MMBioS), linked tothe Pittsburgh University School of Medicine, Bahars Lab. During this sabbatical year, Dra. Ana Ligia studied biomolecules and systems involved in neuronal signaling as: Pink1, PKA, VCP and α-synuclein. This project will allow us to join the knowledge acquired at Bahars Group (Multiscale Modeling, Elastic Network Models) with our experience with Molecular Dynamics, Normal Modes Analysis and Docking. The project will allow our Laboratory keep to research the relationship between structural dynamics of proteins and neurodegenerative diseases. |
|
|
Nanopartículas e ligas metálicas para melhoria de células de combustível a etanol (nanom) Universidade Tecnológica Federal do Paraná Departamento de Ciências da Natureza Coordenador: Vagner Alexandre Rigo Áreas do conhecimento: Engenharias; Física; Química Início da vigência: 01-08-2017 Células combustíveis podem converter combustíveis como o etanol em eletricidade com grande eficiência. Atualmente quase metade da frota de automóveis do Brasil é movida com etanol, fazendo das células de combustíveis a etanol uma área estratégica no país. A oxidação completa do etanol poderia gerar um total de 12 elétrons por molécula (com densidade de energia superior ao hidrogênio ou metanol). Entretanto, os materiais atualmente empregados nos ânodos das células a etanol conseguem fornecer entre 2 e 4 elétrons/molécula, devido a formação de aldeídos. A melhora na eficiência da conversão do etanol em eletrícidade é uma aplicação onde o desenvolvimento de materiais com propriedade melhoradas por design é desejável. Acredita-se que a reação de oxidação do etanol pode ser mais eficiente através do design melhorado de superfícies de Pt em ligas, superfícies com cavidades e nanopartículas. Opta-se por ligas de Au-Pt e Pd-Pt devido a resultados preliminares favoráveis (sistema já abordado pelos autores), onde se almeja avaliar a oxidação do etanol em ligas Au-Pt e Pd-Pt, considerando (i) efeito da dopagem em superfícies de Pt, Au e Pd, (ii) forma e tamanho de superfícies com cavidades, (iii) efeito do tamanho e composição química de nanopartículas core-shell de até 561 átomos, onde é imprescindível o uso intensivo de supercomputação de alto desempenho. |
|
|
Novas perspectivas em investigações mecanísticas de reações orgânicas (letpoc) Universidade Estadual de Campinas Departamento de Ciência Interdisciplinar Coordenador: Rodrigo Antonio Cormanich Áreas do conhecimento: Química Início da vigência: 07-08-2018 Neste projeto de pesquisa é proposta uma nova perspectiva sobre as investigações do mecanismo de reações orgânicas, usando efeitos dinâmicos ao invés da simples e comum teoria do estado de transição (TST - Transition State Theory). Os estudos de caso deste projeto baseiam-se nas investigações mecanicistas de reações multicomponente como a reação de Mannich, Ugi e Biginelli, usando abordagens teóricas e experimentais. Essas reações multicomponente são complexas e amplamente utilizadas em Química Orgânica e são modelos interessantes para testar a TST, que estuda mecanismos de reação baseados apenas em intermediários e de estados de transição. Como alternativa à TST, propomos o uso de métodos sofisticados envolvendo efeitos dinâmicos, considerando todos os movimentos atômicos que transformam os reagentes em produtos. A esse respeito, serão utilizados os efeitos isotópicos cinéticos (KIE) obtidos em abundância natural, um poderoso método experimental para investigação de mecanismo de reação, amplamente aplicado em estudos mecanísticos, mas ainda não empregado por grupos de pesquisa no Brasil. Cálculos teóricos baseados em métodos teóricos ab initio e também usando vários funcionais DFT serão realizados para aplicar o TST. Cálculos de Dinâmica Molecular (MD) serão aplicados para procurar todos os possíveis movimentos/colisões atômicas durante a coordenada de reação, os quais não são possíveis de ser testados usando somente métodos baseados em TST. Espera-se, por meio desse projeto, que alia métodos teóricos de alto nível com resultados experimentais, elucidar mecanismos de reações orgânicas de interesse da comunidade científica e contribuir para otimização das mesmas, aumentando-se rendimentos, melhorando-se procedimentos experimentais e diminuição da formação de subprodutos indesejáveis. |
|
|
Plataforma Computacional Multiusuária para Análises de Bioinformática em Larga Escala, no Apoio à Rede Nacional Estruturante de Bioinformática (pcmrnbio2) Laboratório Nacional de Computação Científica Laboratório de Bioinformática e Unidade de Genômica Computacional Coordenador: Ana Tereza Ribeiro de Vasconcelos Áreas do conhecimento: Ciência da computação; Ciências biológicas Início da vigência: 02-07-2019 O Laboratório de Bioinformática (LABINFO) do LNCC foi fundado em 2000 pelo Ministério da Ciência, Tecnologia e Inovação (MCTI) e consolidou-se como referência na área de bioinformática e biologia computacional no Brasil. Em 2008, o LABINFO ampliou suas atividades com a criação da Unidade de Genômica Computacional Darcy Fontoura de Almeida (UGCDFA) equipada com máquinas de sequenciamento de nova geração, que atende a várias instituições de pesquisa no Brasil e no mundo. Até o momento, a UGCDFA realizou o sequenciamento de mais de 400 genomas (humanos, fungos, parasitos, vírus, bactérias) de interesse na saúde humana, animal e vegetal, bem como vários metagenomas de interesse biotecnológico. O LABINFO/UGCDFA possui as seguintes funções: I) geração, processamento e análise de sequências de nucleotídeos (genomas, metagenomas, transcriptomas, amplicons, exomas e cDNA); II) armazenamento e distribuição para a comunidade científica dos resultados da análise de sequências geradas pela UGCDFA; III) gerenciamento de redes genômicas no Brasil; IV) desenvolvimento de banco de dados biológicos de aplicação em bioinformática; V) desenvolvimento de metodologias matemáticas e computacionais para estudos de genômica comparativa, estrutural e funcional de organismos de interesse biotecnológico, ambiental e agrícola, e de saúde humana; VI) desenvolvimento de softwares de biologia computacional, e.g. o SABIá , o primeiro software para montagem/anotação automática de genomas no Brasil e o Bioinfo-Gateway , um ambiente de plataforma multiusuário para execuções de softwares de bioinformática em ambientes paralelos e distribuídos (CSGrid/SINAPAD) desenvolvido por iniciativa da Rede Nacional Estruturante de Bioinformática (RNBio); VII) capacitação e orientação de dissertações e teses na linha de pesquisa em bioinformática e biologia computacional na Pós-graduação em Modelagem Computacional (LNCC), e colaboradores (UFRJ, Universidade de Lyon1 e Instituto Evandro Chagas); e VIII) formação de recursos humanos especializados em bioinformática e biologia computacional. A RNBio foi criada em 2014 pelo Programa Estruturante do MCTI e instituições com forte tradição em pesquisas de genômica, proteômica e biologia computacional, a saber, o LNCC, o Laboratório Nacional de Biociências (LNBio/CNPEM) e a UFMG. A missão da RNBio é fomentar o desenvolvimento de projetos de pesquisa no formato multicêntrico e a formação de recursos humanos em estudos temáticos envolvendo biologia computacional, e.g. análises de sequenciamento de genomas, transcriptômica, proteômica, biologia de sistemas e interactoma. A RNBio visa também o constante aperfeiçoamento da infraestrutura computacional disponível pelo que atua estreitamente com os Centros Nacionais de Processamento de Alto Desempenho (CENAPAD) estruturados no sistema SINAPAD. Esses centros conseguiram aumentar a capacidade de execução de análises computacionais em muitas instituições no pais. No cerne da RNBio, desenvolveu-se em 2016 o Bioinfo-Gateway que alavancará pesquisas por meio da execução paralela e distribuída dos softwares mais usados pela comunidade de bioinformática, muitos deles considerados altamente dispendiosos em tempo e processamento. Com a interligação entre grupos de pesquisa de diferentes regiões do país em torno de projetos multicêntricos e a troca de experiência entre grupos especialistas nas várias áreas do conhecimento, a RNBio, e suas contribuições em softwares e pesquisas, irá certamente contribuir para o desenvolvimento científico e tecnológico do país. |
|
|
Simulação Computacional Aplicada à Física dos Materiais (scafmat2) Universidade Federal de Uberlândia Instituto de Física Coordenador: Roberto Hiroki Miwa Áreas do conhecimento: Engenharias; Física; Química Início da vigência: 05-06-2019
Atualmente o desenvolvimento de novos (nano)materiais está atrelada ao conhecimento intimo da matéria, isto é, às suas propriedades físicas e químicas em escala atômica. Nesse contexto, a Simulação Computacional, baseado no formalismo da Mecânica Quântica (MQ), tem um papel de destaque na Física dos Materiais. A aplicação da MQ, utilizando-se de métodos de primeiros princípios, permite no estudo bastante realista da estabilidade energética, das propriedades eletrônicas, estruturais e dinâmicas dos materiais, bem como no desing de novos materiais visando diferentes aplicações tecnológicas (engenharia de materiais), como também no estudo de propriedades fundamentais da matéria (ciência básica). Ações que não são excludentes. Nos últimos anos, os isolantes topológicos têm sido foco de diversos estudos relacionados à robustez dos estados (topológicamente) protegidos, frente a contaminação por elementos (átomos) externos, e a sua interação com superfícies formadas por isolantes triviais ou superfícies metálicas; formado-se assim interfaces topológico/trivial ou topológico/metal. Desde a sua síntese, o grafeno tem sido alvo de intensos estudos, não somente as suas propriedades únicas (o grafeno é palco de diversos estudos relacionados às propriedades fundamentais da matéria), como também visando aplicação na engenharia de materiais, em particular no desenvolvimento de sensores e dispositivos (nano)eletrônicos, como o transistores de efeito de campo. As propriedades eletrônicas e estruturais do grafeno (e outros sistemas 2D) são alteradas, em diferentes escalas, devido efeitos de proximidade ou a formação de ligações químicas, com superfícies sólidas; sejam estas metálicas, semicondutoras ou formadas por isolantes topológicos. Isso abre a um leque imenso de possibilidades de funcionalização do grafeno e de outros sistemas 2D (como os dissulfetos metálicos, e mais recentemente o borofano), mediado por uma engenharia de interfaces compostas por “sistemas-2D/superfícies-sólidas”. Superfícies sólidas (cristalinas), sejam elas metálicas ou semicondutoras (triviais ou topológicas), muitas vezes apresentam arranjos atômicos periódicos devido aos processos de reconstrução de forma a reduzir a energia livre da superfície. Tais reconstruções podem servir como guia para a formação de arranjos moleculares auto-organizados sobre superfícies sólidas. De forma simplificada, a formação desses arranjos são mediados por (a) interações entre moléculas e (b) interações molécula-superfície. Uma adequada sinergia entre (a) e (b) irá reger os processos de auto-organização moleculas. Recentemente, essas estruturas moleculares auto-organizadas tem tido um papel importante, por exemplo, na eletrônica molecular (em particular moléculas orgânicas sobre superfícies semicondutoras) e nos processos de catálise (arranjos moleculares sobre superfícies metálicas).
|
|
|
Simulação computacional de materiais multifuncionais: propriedades eletrônicas, estruturais, magnéticas e magnetoelétricas (magmat) Universidade de São Paulo Instituto de Física Coordenador: Helena Maria Petrilli Áreas do conhecimento: Física Início da vigência: 18-06-2019
O complexo acoplamento entre as ordens elétrica e magnética em multiferroicos, denominado acoplamento magnetoelétrico (ME), ainda desafia nossa compreensão e oferece a perspectiva de novas aplicações. No entanto, os compostos ME à temperatura ambiente são muito raros e os ME artificiais de alta qualidade são difíceis de produzir. Os multiferroicos naturalmente estruturados (NLM) oferecem, a esse respeito, uma alternativa inspiradora para obter MEs mais baratos e de alto desempenho. |
|
|
Simulação computacional de nanoestruturas por meio de cálculos de primeiros princípios (simnano) Universidade Federal do Rio de Janeiro Instituto de Física Coordenador: Marcos Gonçalves de Menezes Áreas do conhecimento: Física Início da vigência: 16-10-2019
Neste projeto, utilizaremos cálculos de primeiros princípios do estado da arte para estudar as propriedades eletrônicas, estruturais e ópticas de uma variedade de nanoestruturas. Nosso foco principal serão nanoestruturas de carbono e materiais 2D (sistemas de baixa dimensionalidade em geral), mas também estudaremos o problema de impurezas em semicondutores usuais. Nosso grupo NAMOR/UFRJ (Nanoscience Modeling in Rio) conta com 4 professores, 2 estudantes de pós-graduação e 2 pós-docs, e todos eles estarão envolvidos em projetos com cálculos a serem desenvolvidos no cluster. Destacamos alguns deles abaixo: |
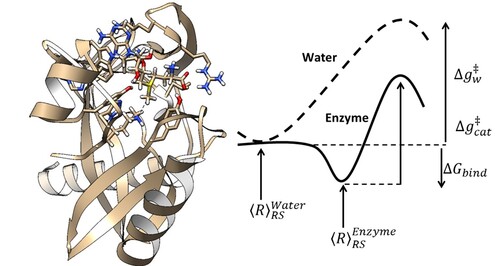
|
Simulação Computacional de Reações Catalisadas por Metiltransferases (scrcm) Universidade Federal do Pará Instituto de Ciências Biológicas Coordenador: Jerônimo Lameira Silva Áreas do conhecimento: Ciências biológicas; Ciências da saúde; Química Início da vigência: 09-07-2018 Apesar do impressionante avanço de software e hardware, o estudo teórico de sistemas compostos por um elevado número de átomos, como é o caso das enzimas, ainda apresenta dificuldades e limitações computacionais. Recentemente, utilizamos o método de mecânica quântica combinado com mecânica molecular (QM/MM) e o método de Dinâmica Molecular para estudar diferentes sistemas biomoleculares (ver lattes http://lattes.cnpq.br/7711489635465954). Nesta proposta, pretende-se utilizar os Supercomputadores Santos Dumont (SDumont/LNCC) e Lobo Carneiro (LoboC/UFRJ) para as aplicações dos referidos métodos no estudo do mecanismo catalítico das enzimas catecol-metiltransferase (COMT), fosfoetanolamina metiltransferase (PfPMT) e Proteína histona metiltransferase (PKMT). Estes métodos são usados por importantes grupos de pesquisa bem como por empresas para propor bioligantes ou inibidores de alvos terapêuticos. Desta forma, pretende-se produzir informações científicas que podem levar ao entendimento da origem do poder catalítico destas enzimas. As informações fornecidas pelos cálculos teóricos podem ser futuramente utilizadas por experimentais os quais podem sintetizar e avaliar a atividade biológica de novas moléculas. Vale ressaltar que esta proposta visa dar continuidade à projetos em andamento no Laboratório de Planejamento e Desenvolvimento de Fármaco da Universidade Federal do Pará, dos quais pode-se destacar um projeto aprovado na CHAMADA UNIVERSAL-MCTI/CNPQ No 14/2014 na faixa A (441922/2014-7) e bolsa de Produtividade em Pesquisa - PQ 2015 - Nível 2 (Vigência: 01/03/2016 - 28/02/2019). Em termos de infraestrutura, o proponente desenvolve pesquisa no Laboratório de Planejamento e Desenvolvimento de Fármaco da Universidade Federal do Pará o qual possui um cluster para realização das simulações computacionais. No entanto, atualmente, a infraestrutura do grupo é utilizada por 11 (onze) alunos de doutorado, 12 (doze) alunos de mestrado e 6 (seis) alunos de graduação, o que tem demandado maior poder computacional. Desta forma, a utilização dos supercomputadores em questão poderia auxiliar no andamento das atividades já mencionadas. |
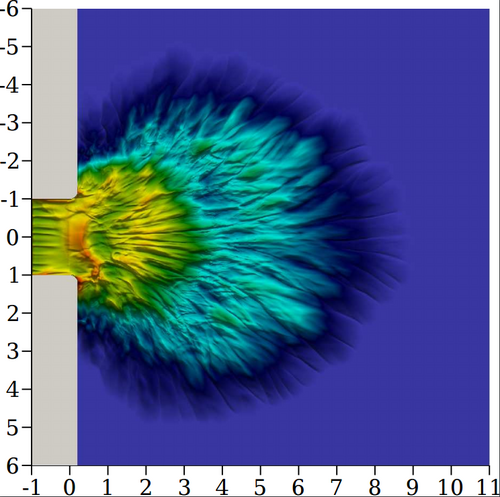
|
Simulação de alta performance de processos deposicionais devido a correntes gravitacionais (DNSDAM) Pontifícia Universidade Católica do Rio Grande do Sul Faculdade de Engenharia - FENG Coordenador: Jorge Hugo Silvestrini Áreas do conhecimento: Ciência da computação; Engenharias; Geociências Início da vigência: 18-05-2017 O projeto visa, através de simulações numérica de alta fidelidade, caracterizar geometricamente os depósitos gerados por correntes gravitacionais hipopicnais e hiperpicnais com transporte de sedimentos. A metodologia usada para resolver as equações governantes é a Simulação Numérica Direta (DNS) a qual permite calcular com grande exatidão, todas as escalas do escoamento turbulento e, por tanto, reproduzir de forma acurada, as diferentes instabilidades que ocorrem no processo. Dos resultados obtidos usando DNS, pode-se interpretar a complexa organização espacial e temporal provocada pela frente da corrente caracterizadas por estruturas de lobos e fendas e a influência que estas apresentam na formação de canais sedimentares. Um ponto a destacar é a distribuição espacial tridimensional de camadas de diferentes granulometrias, informação de extrema utilidade para identificar reservatórios O objetivo final é compreender como são formados os depósitos sedimentares e sua evolução temporal e espacial, informações essenciais para a exploração de petróleo e gás. |

|
Simulação de combustão de spray de etanol para aplicação em motores de combustão interna (motoretanol2) Universidade de São Paulo Escola Politécnica Coordenador: Guenther Carlos Krieger Filho Áreas do conhecimento: Engenharias Início da vigência: 11-06-2019
A presente proposta contempla a continuação do projeto MotorEtanol, que faz parte de uma iniciativa da Fundação de Amparo à Pesquisa do Estado de São Paulo (Fapesp) em conjunto com a empresa Peugeot Citröen do Brasil Automóveis Ltda (PCBA), que financiam o Centro de Pesquisa em Engenharia "Prof. Urbano Ernesto Stumpf (http://www.bv.fapesp.br/pt/auxilios/84719/estudo-conceitual-de-um-motor-avancado-a-etanol). Pesquisadores de várias universidades, entre elas a Unicamp, o ITA, a USP e o Instituto Mauá de Tecnologia (IMT) fazem parte do Centro de Pesquisas. O Centro destina-se a estudar o conceito de um motor avançado movido a etanol, que explore as especificidades e características positivas do etanol como combustível. O resultado esperado é uma proposta conceitual para apresentar um motor com um melhor desempenho de etanol e, simultaneamente, uma melhor eficiência. |
|
Simulação Multiescala de Problemas da Engenharia do Petróleo Usando o Método MHM (mhm) Laboratório Nacional de Computação Científica Coordenação de Matemática Aplicada e Computacional Coordenador: Frederic Valentin Áreas do conhecimento: Ciência da computação; Engenharias; Matemática Início da vigência: 28-06-2016 Este projeto propõe analisar computacionalmente algoritmos numéricos derivados de uma nova família de métodos de elementos finitos multi-escalas chamada de MHM – Multiscale Hybrid-Mixed. Os métodos MHM são adaptados à resolução de modelos tridimensionais com múltiplas escalas ou com grandes contrastes, caracterizando-se por alta ordem de precisão (baixas taxas de erro) e por incorporar a granularidade e heterogeneidade das novas gerações de arquiteturas massivamente paralelas. Pretende-se aplicar a nova metodologia à simulação de problemas da engenharia de petróleo no âmbito dos projetos HPC4e e HOMAR, com particular interesse em modelos provenientes das áreas de sísmica e de reservatório de petróleo. O objetivo final é inferir/demonstrar a capacidade do algoritmo MHM em resolver modelos com bilhões de graus de liberdade em poucos minutos com imediato impacto na área de engenharia de petróleo. A ultima década foi marcada por um grande desenvolvimento de computadores com arquiteturas massivamente paralelas. As novas arquiteturas baseiam-se na multiplicação do número de processadores (agrupados em cores) com baixa/média velocidade e capacidade de estocagem. Esse novo paradigma implica na revisão do que se espera dos simuladores computacionais. Embora a precisão e robustez dos métodos numéricos continuem sendo atributos fundamentais para a qualidade das simulações computacionais, estes devem ser capazes de naturalmente incorporarem as características intrínsecas das novas gerações de arquiteturas, alem de serem tolerantes a falhas. De fato, falhas durante simulações computacionais de grande vulto são uma certeza neste cenário. Consequentemente, os novos métodos numéricos precisam necessariamente induzir algoritmos assíncronos e que evitem ao máximo comunicações entre processadores. Os métodos numéricos baseados na estratégia de “dividir para conquistar” preenchem tais imperativos arquiteturais, os quais não estão presentes na construção dos métodos numéricos clássicos. De fato, dividir a computação associada a simulações de “altíssimo porte” em um imenso conjunto de problemas independentes de pequeno tamanho apresenta-se como a estratégia desejável. Como resultado, limita-se o impacto das falhas de hardware e tira-se proveito da localidade temporal e espacial das simulações. O método MHM se adequa perfeitamente a esse cenário. Para tanto, dada uma partição (grossa) do domínio, o método MHM relaxa a continuidade da solução que é imposta através de multiplicadores de Lagrange (hibridização). Demonstra-se que a solução exata dos problemas de valores de contorno e/ou de valor inicial incorporam os aspectos multi-escalas e de alto-contraste através de problemas definidos elemento a elemento com condições de contorno dependentes dos multiplicadores de Lagrange. Discretizando-se esse modelo obtêm-se o método MHM, composto de uma formulação global definida no esqueleto da partição, e de uma coleção de problemas locais dirigidos pelos dados do problema e pelos multiplicadores de Lagrange aproximados por polinômios de alta ordem. Ao final, a nova família de métodos MHM possui as seguintes propriedades: (i) Estabilidade e alta ordem de aproximação dirigidos pela aproximação nas faces da partição grossa do domínio; (ii) Inclusão de processos de “upscaling” locais e independente via funções de base, sem a necessidade de “oversampling”; (iii) Produz campos localmente conversativos; (iv) Induz adaptabilidade temporal e espacial via um estimador de erro a posteriori. |
|
|
Simulação Multiescala de Problemas da Engenharia do Petróleo Usando o Método MHM (mhm2) Laboratório Nacional de Computação Científica Coordenação de Matemática Aplicada e Computacional Coordenador: Frederic Valentin Áreas do conhecimento: Ciência da computação; Engenharias; Matemática Início da vigência: 17-01-2020
Este projeto propõe analisar computacionalmente algoritmos numéricos derivados de uma nova família de métodos de elementos finitos multi-escalas chamada de MHM – Multiscale Hybrid-Mixeds métodos MHM são adaptados à resolução de modelos tridimensionais com múltiplas escalas ou com grandes contrastes, caracterizando-se por alta ordem de precisão (baixas taxas de erro) e por incorporar a granularidade e heterogeneidade das novas gerações de arquiteturas massivamente paralelas. Pretende-se aplicar a nova metodologia à simulação de problemas da engenharia de petróleo no âmbito dos projetos HPC4e e HOMAR (detalhes sobre os mesmos encontram-se adiante na proposta), com particular interesse em modelos provenientes das áreas de sísmica e de reservatório de petróleo. O objetivo final é inferir/demonstrar a capacidade do algoritmo MHM em resolver modelos com bilhões de graus de liberdade em poucos minutos com imediato impacto na área de engenharia de petróleo. |
|
Simulações 'high throughput' para o design e descoberta de materiais (mat_design) Universidade Federal do ABC Centro de Ciências Naturais e Humanas Coordenador: Gustavo Martini Dalpian Áreas do conhecimento: Engenharias; Física Início da vigência: 15-05-2019
A modelagem computacional é uma técnica poderosa para o desenvolvimento de novos materiais. Esta área teve um avanço enorme nas últimas décadas, atingindo recentemente sua plena maturidade. Este avanço fez com que novas fronteiras se abrissem para o desenvolvimento da área, e uma das vertentes mais fortes foca na produção e no tratamento de grandes quantidades de dados. Chegamos ao momento em que a Simulação Computacional de Materiais ande em direção à área de Big Data [1]. Hoje no Brasil há poucos grupos trabalhando nesta área, apesar dos grandes esforços da comunidade internacional neste sentido. O objetivo deste projeto é o design e descoberta de novos materiais através da realização de simulações computacionais de forma 'high throughput', mapeando todo o espaço de fases desses materiais, e procurando por compostos com propriedades específicas. Para a realização dessas simulações, acoplamos um código padrão baseado na Teoria do Funcional da Densidade (DFT), com scripts que permitem a realização de um grande número de simulações ao mesmo tempo. Esse tipo de metodologia muitas vezes é referido como 'Inverse Design'[2]. |
|
|
Simulações computacionais ab initio em materiais nanoestruturados: propriedades estruturais, eletrônicas e de transporte (nanocomp) Universidade Federal de Goiás Instituto de Física Coordenador: Andreia Luisa da Rosa Áreas do conhecimento: Física; Química Início da vigência: 18-07-2019 Através de uma engenharia em escala nanométrica, átomos e/ou moléculas sintetizados para específicas funcionalidades podem ser manipulados de acordo com o interesse. Adicionalmente, aparecem fenômenos fundamentalmente novos, como a quantização da condutância. Propomos aqui um projeto de pesquisa focado em simulações ab initio, aplicadas à sistemas nanoestruturados, mas com grande apelo experimental. O projeto visa efetuar a caracterização das propriedades mecânicas, eletrônicas e de transporte de nanoestruturas metálicas, entre elas: nanofios metálicos, junções bimetálicas e pequenos clusters. Estes sistemas metálicos são sistemas ideais para o uso por exemplo em produção de hidrogênio e conversores catalíticos. Assim, o desafio atual é desenvolver materiais pequenos, baratos e leves. Nesse sentido, a simulação computacional pode fornecer resultados quantitativos e pode servir como uma alternativa para experimentos que são muito difíceis ou caros de serem realizados. Nesta categoria incluem-se nanoestruturas metálicas bem como a funcionalização dos mesmos através de dopagem e ligantes orgânicos. Do ponto de vista teórico os problemas abordados exigem combinações de vários métodos para se obter resultados precisos que permitam fazer previsões que possam ser usada a orientação de medidas experimentais, ou que favoreça um melhor entendimento dos mecanismos físicos envolvidos. |
|
Simulações de alta-fidelidade alta fidelidade e modelagem de ordem reduzida de escoamentos não-estacionários estacionários envolvendo turbulência e instabilidades hidrodinâmicas (simturb) Universidade Estadual de Campinas Faculdade de Engenharia Mecânica - Departamento de Energia Coordenador: William Roberto Wolf Áreas do conhecimento: Engenharias Início da vigência: 07-06-2019 Neste projeto pretendemos realizar simulações de alta-fidelidade de escoamentos não-estacionários e desenvolver modelos de ordem reduzida através de técnicas de deep learning e identificação de sistemas dinâmicos não-lineares. Focaremos no estudo de escoamentos turbulentos ou envolvendo transição que ocorrem em aplicações de engenharia aeroespacial. As configurações investigadas incluem aerofólios com camadas-limite coladas, aerofólios em estol dinâmico, camadas-limite supersônicas e escoamentos em cavidades cilíndricas com rotação. As aplicações desses estudos são diversas incluindo o projeto de pás de turbina e rotores de helicópteros, motores de aeronaves supersônicas além de compressores de turbinas aeronáuticas. Simulações de grandes escalas, LES, e simulações numéricas diretas, DNS, são as abordagens escolhidas para serem aplicadas neste projeto. Estas abordagens serão aplicadas com o objetivo de resolver mecanismos físicos transientes nos escoamentos. Para os estudos de escoamentos sobre aerofólios e em canais, esquemas numéricos compactos de diferenças finitas com alta resolução serão empregados usando LES e DNS para a solução de estruturas turbulentas e de instabilidade com o mínimo possível de dissipação e dispersão numérica. Os problemas envolvendo cavidades serão resolvidos por métodos de elementos espectrais de alta ordem. Ambas as metodologias irão empregar malhas estruturadas apropriadas para a resolução de camadas limite sobre as configurações de aerofólio, canais e cavidades. Os resultados das simulações numéricas serão utilizados para se treinar modelos de ordem reduzida utilizando técnicas de projeção de Galerkin e outras baseadas em redes neurais profundas. Atualmente o desenvolvimento de modelos de ordem reduzida é um tópico bastante promissor pois visa reduzir o custo computacional das simulações de alta-fidelidade sendo ao mesmo tempo capaz de reproduzir as principais características físicas dos escoamentos. O atual grupo de pesquisa da Unicamp vem se destacando internacionalmente no desenvolvimento de modelos de ordem reduzida e, também, na aplicação de técnicas de decomposição modal de escoamentos que servem para reduzir a dimensionalidade dos problemas em questão. Este projeto será desenvolvido em parceria entre a Unicamp e a UFPR e os recursos computacionais do computador SDumont serão imprescindíveis para a conclusão do mesmo. |

|
Simulações e análise de dados de astropartículas (astroparti) Universidade de São Paulo Departamento de Ciência Interdisciplinar Coordenador: Luiz Vitor de Souza Filho Áreas do conhecimento: Astrofísica; Física Início da vigência: 26-10-2016 O Observatório Pierre Auger é o maior experimento de astrofísica de partículas em operação. O Observatório vem tomando dados nos últimos 10 anos e vem produzindo uma revolução no nosso conhecimento à respeito da produção das partículas mais energéticas do Universo. Desde a sua inauguração, o Observatório já produziu mais de 80 artigos em revista científicas de alto impacto com um total de mais de 7500 citações. A colaboração brasileira que participa e contribui com estas publicações conta com mais de 20 doutores em 12 instituções de pesquisa espalhadas pelo Brasil. Nossa participação é bastante significativa no Observatório. Temos liderado várias forças tarefas na organização internacional do experimento (17 países), produzindo um número significante de doutorados e envolvido a indústria nacional. Neste quisito, várias indústrias nacionais participaram da construção de equipamentos para o Observatório agregando conhecimento e inovação ao parque industrial brasileiro. Recentemente, o acordo internacional de operação do Observatório foi extendido até 2024 e iniciamos uma etapa de melhorias das instalações dos detectores da qual as universidades e industrias nacionais continuarão participando. Os próximos 10 anos de operação do Observatório Pierre Auger não serão menos produtivos do que a última década. A análise de dados do Observatório depende de simulação detalhada de vários fenômenos físicos e dos detectores. A propagação da partícula da fonte até a Terra, a interação da partícula na atmosféra e a interação das partículas com os detectores precisam ser minuciosamente descritas para que os dados medidos possam ser interpretados. Como pode ser comprovado nos CV Lattes dos proponentes, os pesquisadores brasileiros neste projeto tem experiência no desenvolvimento de softwares para resolver esses problemas de física e transformá-los em conhecimento científico de primeira magnitude. A colaboração brasileira no Observatório Pierre Auger não possue recursos computacionais específicos para a realiação de sua pesquisa. Os resultados anteriormente conseguidos foram alcançados em pequenos conjuntos de processadores (< 50) o que demanda vários meses de processamento para resultados muitas vezes estatisticamente insatisfatórios. Por vezes, colaborações com instituções no exterior tem garantido acesso mínimo a recursos computacionais. Desta forma, apresentamos este projeto de uso do supercomputador Santos Dumont (SDumont). Acreditamos que nossa pesquisa se benificiará imensamente do acesso ao SDumont pois permitirá aos pesquisadores e alunos, atingirem um nível de precisão inédito nos seus resultados em curto período de tempo. |

|
Simulações e cálculos numéricos aplicados à aceleradores para o Centro Nacional de Pesquisa em Energia e Materiais (CNPEM) (sirius) Centro Nacional de Pesquisa em Energia e Materiais Grupo de Proteção Radiológica Coordenador: Isabela Castro de Moraes Áreas do conhecimento: Física Início da vigência: 21-09-2016
O Brasil terá um novo acelerador de elétrons, o Sírius, o segundo síncrotron de 4a geração a ser construído no mundo, e o de mais alto brilho, que está sendo construído pelo Laboratório Nacional de Luz Síncrotron (LNLS) em Campinas.
Assim como o UVX, que foi o primeiro acelerador síncrotron construído pelo LNLS e o primeiro do hemisfério Sul, o Sirius será disponibilizado para toda a comunidade nacional e internacional, para a realização de pesquisas em física, química, medicina, biologia, agricultura, ciências de materiais, farmacologia, geofísica e muitas outras áreas.
Um dos maiores diferenciais do Sirius em relação ao estado da arte mundial dos síncrotrons é sua baixíssima emitância (parâmetro que caracteriza quão compacto é o espaço de fase dos elétrons no acelerador, medido em nanômetros-radianos). Tal diferencial tornará o Sirius um líder mundial, com a menor emitância natural dentre os síncrotrons no mundo. Assim, os feixes de radiação eletromagnética produzidos nesse acelerador, iluminarão microscópios (linhas de luz) que farão imagens tridimensionais de materiais, com resoluções espaciais nanométricas, em condições de operação, e com os mais diversos tipos de contraste permitidos pelas interações da radiação eletromagnética com a matéria.
Para blindar a radiação sincrotron e o bremsstrahlung presentes nas estações experimentais (cabanas) será necessário utilizar-se de espessuras adequadas de materiais apropriados para esse fim. A análise radiológica da blindagem para o acelerador leva em consideração os seguintes componentes:
|
|
|
Simulações moleculares em multiescala aplicadas na separação e detecção do gás natural por membranas (p41) Universidade de São Paulo Instituto de Física Coordenador: Alexsandro Kirch Áreas do conhecimento: Engenharias; Física; Química Início da vigência: 26-11-2019 O desafio permanente na indústria do gás é obter membranas que exibam simultaneamente alta seletividade e permeabilidade para a separação eficiente do gás natural. Além disso, há o interesse em desenvolver dispositivos de detecção mais baratos e sensíveis capazes de identificar os componentes de um determinado gás. Essa tecnologia é particularmente importante para detectar o sulfeto de hidrogênio em ambientes de exploração de petróleo, uma vez que ele é altamente tóxico. Tais dispositivos podem ser projetados para obter metano limpo na indústria do gás e avaliar a eficiência do processo de separação. O surgimento da nanotecnologia revelou um novo conjunto de possibilidades que podem melhorar esses processos significativamente, uma vez que ela permitiria manipular e detectar moléculas individuais. A nanociência pode ser aplicada na indústria de petróleo e gás em processos que vão do upstream até o downstream. Atualmente há o interesse de obter soluções para os desafios encontrados na separação e detecção do gás natural através da nanociência. Nesse contexto, a compreensão dos efeitos do gás natural nas propriedades eletrônicas e de transporte dos materiais nano estruturados é fundamental para o desenvolvimento de futuras aplicações em sensores e dispositivos de separação mais eficientes. Membranas bidimensionais e nanotubos baseados em carbono, nitreto de boro, dissulfetos de molibdênio (MoS2), titânio (TiS2) e tungstênio (WS2) são sistemas promissores para o desenvolvimento de tais dispositivos. Nesse projeto, propomos investigar o efeito dos principais componentesdo gás natural e sua mistura nas propriedades eletrônicas e de transporte desses nanosistemas com o objetivo de compreender os processos de detecção e separação de gases. Para realizar esse estudo, utilizaremos uma abordagem multi-escala em nível molecular que combina simulações de dinâmica molecular com cálculos de primeiros princípios de transporte eletrônico. Essa pesquisa pode levar à otimização dos dispositivos, resultando em processos mais eficientes de detecção e separação do gás natural. |
|
|
Simulações Moleculares em Multiescala com Aplicações em Recuperação Melhorada de Petróleo: Baixa Salinidade em Carbonatos (eorlssampa) Universidade de São Paulo Instituto de Física Coordenador: Caetano Rodrigues Miranda Áreas do conhecimento: Ciência dos materiais; Física; Química Início da vigência: 27-11-2019
Recentemente, observou-se que a injeção de água de baixa salinidade leva a uma melhoria nos processos de recuperação melhorada de petróleo (EOR), com a vantagem de ser uma tecnologia ambientalmente amigável e utilizar insumos in-situ. O conhecimento dos fundamentos desse fenômeno permite a proposição de uma solução salina ótima, melhorando significativamente a produção de petróleo. Entretanto, o entendimento dos mecanismos moleculares adjacentes a esse fenômeno não apenas é desconhecido, mas é também dependente do tipo de material geológico envolvido. A aplicação de simulações computacionais com uma metodologia de múltiplas escalas, tais como primeiros princípios e dinêmica molecular clássica, possibilita caracterizar as reações geoquímicas de interesse, bem como determinar as propriedades termodinâmicas e cinéticas envolvidas nessas reações intra-aquosas, como os processos de dissolução e precipitação de minerais, titulação, troca de íons e a alteração da molhabilidade, que no caso dos carbonatos não são conhecidas nas condições de reservatório. |
|
Simulando a hidrodinâmica do gás no interior de galáxias anãs (hd3ddsph2) Universidade Cruzeiro do Sul Núcleo de Astrofísica Teórica Coordenador: Anderson Caproni Áreas do conhecimento: Astronomia Início da vigência: 27-06-2019 Galáxias esferoidais anãs estão entre os objetos menos luminosos e mais maciços (dominados pela matéria escura) do nosso universo local conhecidos até o presente momento. Uma das características mais marcantes dessas galáxias é a ausência quase completa de gás neutro em seus interiores, com as causas que produziram tal remoção sendo ainda matéria de debate na literatura científica. Mecanismos internos (associados à atividade estelar nessas galáxias, por exemplo, supernovas) e de origem externa (relacionados à interação do meio interestelar das galáxias com perturbadores externos a essas, por exemplo, pressão de arrasto produzida pelo meio intergaláctico) são usualmente evocados para explicar a ausência de gás no interior das galáxias esferoidais anãs nos dias atuais. Devido à natureza não linear na interação desses mecanismos físicos com o gás nessas galáxias, bem como a ausência de simetria espacial nessas interações, se faz necessário o uso de simulações hidrodinâmicas em três dimensões para uma descrição mais apropriada do comportamento dinâmico do gás ao longo da história de evolução desses sistemas. Nesse contexto, o objetivo principal do presente projeto é estudar, do ponto de vista hidrodinâmico, como as galáxias esferoidais anãs perderam seus respectivos conteúdos gasosos ao longo de sua história evolutiva priorizando os mecanismos físicos de remoção via supernovas do tipo II e Ia, remoção por pressão de arrasto induzida pelo movimento dessas galáxias através do meio intergaláctico, e o outflow de buracos negros de massa intermediária no centro dessas galáxias. Será analisado ainda o papel do perfil de matéria escura na taxa da perda de massa nesses objetos. O fato de analisarmos sistemas com dimensões físicas da ordem de milhares de persecs com resoluções espaciais de apenas alguns parsecs, impõe um grande desafio numérico que pode ser superado apenas com o uso de supercomputadores como o pleiteado neste projeto. |
|
Spin dynamics in magnetic topological insulators: implications for quantum and classical devices (sdtopol) Universidade Federal Fluminense Instituto de física Coordenador: Antonio Tavares da Costa Áreas do conhecimento: Física Início da vigência: 25-06-2018 This project aims at exploring the potential of magnetic topological insulators as building blocks for classical communication devices of nanoscopic dimensions or quantum communication devices. The first part of the project will assess their possible usefulness as quantum and classical data buses. The second part will deal with their properties as nano emitters of electromagnetic radiation in the terahertz frequency range. Topological insulators have unique electronic properties stemming from their peculiar electronic structure: a set of topologically protected edge states and an insulating bulk. The edge states can be used as practically lossless charge channels. They also have unique spin transport properties, such as a ground state spin current. The combination between topological insulators and magnetic adatoms or clusters can be used to store, transmit and manipulate information, both classically and quantum mechanically. Carefully chosen magnetic adatoms may be used to provide the needed coupling with the protected edge states without disturbing significantly their transport properties. They can also serve as qubits or classical bits themselves: the large spin-orbit coupling present in the topological insulator induces large magnetocrystalline anisotropy in the magnetic adatom, making its magnetization direction stable at finite temperatures. The same strong anisotropy renders each magnetic adatom a nano oscillator with a natural oscillation frequency in the terahertz range. If a number of magnetic adatoms is deposited onto a topological insulator, the edge states will couple their magnetizations and can, eventually, lead to synchronized or phase-locked nano oscillators. Those are, in principle, useful resources for building medium and long range miniaturized communication devices. By formulating realistic models for the electronic structure of the magnetic topological insulators we will be able to evaluate the energy dissipation rate of the relevant processes quantitatively. The study of the thermodynamics of spin currents and spin excitations will allow us to devise energy efficient data buses and nano emitters. It will also shed light onto the fundamental features of spin relaxation, which is currently a subject of intense debate. Our results should be useful for guiding experimental attempts at building energy efficient computational devices. |

|
TopSim: Um framework baseado em plugins para análise numérica em larga escala. (TopSim) Pontifícia Universidade Católica do Rio de Janeiro Informática Coordenador: Waldemar Celes Filho Áreas do conhecimento: Ciência da computação; Engenharias Início da vigência: 17-05-2017 O principal objetivo desta pesquisa é o uso de computação híbrida, distribuída e paralela (futuramente também em GPU), aplicada à análise numérica pelo Método dos Elementos Finitos (MEF) para a solução de problemas de otimização topológica. Uma das metas é realizar a maior simulação de otimização topológica já feita até o momento, com 1 bilhão de incógnitas em um sistema linear de equações. O projeto pretende dar continuidade à colaboração com o Prof. Glaucio Paulino da Georgia Tech University, responsável por disponibilizar o supercomputador Blue Waters, onde uma primeira versão do código já foi implementada e testada, com o apoio do CNPq. Ressalta-se que a colaboração com o Prof. Paulino e sua equipe já gerou a publicação de dois trabalhos em periódicos: “Topology optimization using polytopes”, Journal of Computational Physics; e “PolyTop++: An efficient alternative for serial and parallel topology optimization on CPUs & GPUs”, Journal of Structural and Multidisciplinary Optimization. A continuação da pesquisa é fundamental para o desenvolvimento do sistema em supercomputadores, garantindo eficiência com modelos de larga escala. Uma das principais áreas de interesse deste trabalho é a solução de problemas de otimização topológica com malhas da ordem de centenas de milhões de elementos. O método de otimização topológica é usado em problemas que buscam a distribuição ótima da quantidade de material no interior de um domínio submetido a um conjunto de carregamentos e condições de contorno. Este método tem sido aplicado em diferentes áreas, tais como: bioengenharia, na qual pacientes em reconstrução facial podem fazer uma substituição óssea com sua forma e função otimizada; no projeto de asas de avião, com o mínimo de material e o máximo de resistência às condições de uso; e no projeto de peças automotivas e mecânica dos fluidos. |
|
|
Utilização do SD por parte do experimento LHCB do CERN e seu grupo brasileiro (lhcb) Centro Brasileiro de Pesquisas Físicas Coordenação de Física de altas Energias Coordenador: Renato Santana Áreas do conhecimento: Física; Matemática Início da vigência: 31-01-2017
|